Mohammad Amin Ghanavati, Soroush Ahmadi and Sohrab Rohani
{"title":"A machine learning approach for the prediction of aqueous solubility of pharmaceuticals: a comparative model and dataset analysis†","authors":"Mohammad Amin Ghanavati, Soroush Ahmadi and Sohrab Rohani","doi":"10.1039/D4DD00065J","DOIUrl":null,"url":null,"abstract":"<p >The effectiveness of drug treatments depends significantly on the water solubility of compounds, influencing bioavailability and therapeutic outcomes. A reliable predictive solubility tool enables drug developers to swiftly identify drugs with low solubility and implement proactive solubility enhancement techniques. The current research proposes three predictive models based on four solubility datasets (ESOL, AQUA, PHYS, OCHEM), encompassing 3942 unique molecules. Three different molecular representations were obtained, including electrostatic potential (ESP) maps, molecular graph, and tabular features (extracted from ESP maps and tabular Mordred descriptors). We conducted 3942 DFT calculations to acquire ESP maps and extract features from them. Subsequently, we applied two deep learning models, EdgeConv and Graph Convolutional Network (GCN), to the point cloud (ESP) and graph modalities of molecules. In addition, we utilized a random forest-based feature selection on tabular features, followed by mapping with XGBoost. A t-SNE analysis visualized chemical space across datasets and unique molecules, providing valuable insights for model evaluation. The proposed machine learning (ML)-based models, trained on 80% of each dataset and evaluated on the remaining 20%, showcased superior performance, particularly with XGBoost utilizing the extracted and selected tabular features. This yielded average test data Mean Absolute Error (MAE), Root Mean Squared Error (RMSE), and <em>R</em>-squared (<em>R</em><small><sup>2</sup></small>) values of 0.458, 0.613, and 0.918, respectively. Furthermore, an ensemble of the three models showed improvement in error metrics across all datasets, consistently outperforming each individual model. This Ensemble model was also tested on the Solubility Challenge 2019, achieving an RMSE of 0.865 and outperforming 37 models with an average RMSE of 1.62. Transferability analysis of our work further indicated robust performance across different datasets. Additionally, SHAP explainability for the feature-based XGBoost model provided transparency in solubility predictions, enhancing the interpretability of the results.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 10","pages":" 2085-2104"},"PeriodicalIF":6.2000,"publicationDate":"2024-09-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d4dd00065j?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d4dd00065j","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
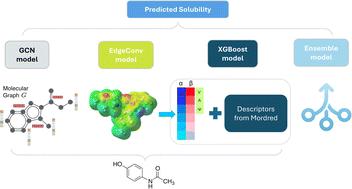
The effectiveness of drug treatments depends significantly on the water solubility of compounds, influencing bioavailability and therapeutic outcomes. A reliable predictive solubility tool enables drug developers to swiftly identify drugs with low solubility and implement proactive solubility enhancement techniques. The current research proposes three predictive models based on four solubility datasets (ESOL, AQUA, PHYS, OCHEM), encompassing 3942 unique molecules. Three different molecular representations were obtained, including electrostatic potential (ESP) maps, molecular graph, and tabular features (extracted from ESP maps and tabular Mordred descriptors). We conducted 3942 DFT calculations to acquire ESP maps and extract features from them. Subsequently, we applied two deep learning models, EdgeConv and Graph Convolutional Network (GCN), to the point cloud (ESP) and graph modalities of molecules. In addition, we utilized a random forest-based feature selection on tabular features, followed by mapping with XGBoost. A t-SNE analysis visualized chemical space across datasets and unique molecules, providing valuable insights for model evaluation. The proposed machine learning (ML)-based models, trained on 80% of each dataset and evaluated on the remaining 20%, showcased superior performance, particularly with XGBoost utilizing the extracted and selected tabular features. This yielded average test data Mean Absolute Error (MAE), Root Mean Squared Error (RMSE), and R-squared (R2) values of 0.458, 0.613, and 0.918, respectively. Furthermore, an ensemble of the three models showed improvement in error metrics across all datasets, consistently outperforming each individual model. This Ensemble model was also tested on the Solubility Challenge 2019, achieving an RMSE of 0.865 and outperforming 37 models with an average RMSE of 1.62. Transferability analysis of our work further indicated robust performance across different datasets. Additionally, SHAP explainability for the feature-based XGBoost model provided transparency in solubility predictions, enhancing the interpretability of the results.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
