Yichen Si, ChangHee Lee, Yongha Hwang, Jeong H. Yun, Weiqiu Cheng, Chun-Seok Cho, Miguel Quiros, Asma Nusrat, Weizhou Zhang, Goo Jun, Sebastian Zöllner, Jun Hee Lee, Hyun Min Kang
{"title":"FICTURE: scalable segmentation-free analysis of submicron-resolution spatial transcriptomics","authors":"Yichen Si, ChangHee Lee, Yongha Hwang, Jeong H. Yun, Weiqiu Cheng, Chun-Seok Cho, Miguel Quiros, Asma Nusrat, Weizhou Zhang, Goo Jun, Sebastian Zöllner, Jun Hee Lee, Hyun Min Kang","doi":"10.1038/s41592-024-02415-2","DOIUrl":null,"url":null,"abstract":"Spatial transcriptomics (ST) technologies have advanced to enable transcriptome-wide gene expression analysis at submicron resolution over large areas. However, analysis of high-resolution ST is often challenged by complex tissue structure, where existing cell segmentation methods struggle due to the irregular cell sizes and shapes, and by the absence of segmentation-free methods scalable to whole-transcriptome analysis. Here we present FICTURE (Factor Inference of Cartographic Transcriptome at Ultra-high REsolution), a segmentation-free spatial factorization method that can handle transcriptome-wide data labeled with billions of submicron-resolution spatial coordinates and is compatible with both sequencing-based and imaging-based ST data. FICTURE uses the multilayered Dirichlet model for stochastic variational inference of pixel-level spatial factors, and is orders of magnitude more efficient than existing methods. FICTURE reveals the microscopic ST architecture for challenging tissues, such as vascular, fibrotic, muscular and lipid-laden areas in real data where previous methods failed. FICTURE’s cross-platform generality, scalability and precision make it a powerful tool for exploring high-resolution ST. FICTURE is a segmentation-free approach for identifying tissue architecture in spatial transcriptomics data. FICTURE is compatible with both imaging-based and sequencing-based methods and is uniquely suited for handling the largest available datasets.","PeriodicalId":18981,"journal":{"name":"Nature Methods","volume":"21 10","pages":"1843-1854"},"PeriodicalIF":32.1000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Methods","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41592-024-02415-2","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
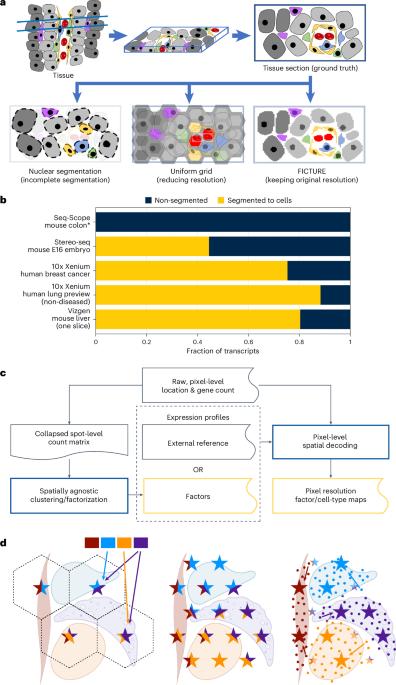
Spatial transcriptomics (ST) technologies have advanced to enable transcriptome-wide gene expression analysis at submicron resolution over large areas. However, analysis of high-resolution ST is often challenged by complex tissue structure, where existing cell segmentation methods struggle due to the irregular cell sizes and shapes, and by the absence of segmentation-free methods scalable to whole-transcriptome analysis. Here we present FICTURE (Factor Inference of Cartographic Transcriptome at Ultra-high REsolution), a segmentation-free spatial factorization method that can handle transcriptome-wide data labeled with billions of submicron-resolution spatial coordinates and is compatible with both sequencing-based and imaging-based ST data. FICTURE uses the multilayered Dirichlet model for stochastic variational inference of pixel-level spatial factors, and is orders of magnitude more efficient than existing methods. FICTURE reveals the microscopic ST architecture for challenging tissues, such as vascular, fibrotic, muscular and lipid-laden areas in real data where previous methods failed. FICTURE’s cross-platform generality, scalability and precision make it a powerful tool for exploring high-resolution ST. FICTURE is a segmentation-free approach for identifying tissue architecture in spatial transcriptomics data. FICTURE is compatible with both imaging-based and sequencing-based methods and is uniquely suited for handling the largest available datasets.
空间转录组学(ST)技术已发展到能以亚微米分辨率对大面积区域进行全转录组基因表达分析。然而,高分辨率 ST 的分析往往受到复杂组织结构的挑战,现有的细胞分割方法因细胞大小和形状不规则而难以实现,而且缺乏可扩展到全转录组分析的无分割方法。在这里,我们提出了 FICTURE(超高分辨制图转录组因式推断),这是一种免分割空间因式分解方法,可处理标有数十亿亚微米分辨率空间坐标的全转录组数据,并与基于测序和成像的 ST 数据兼容。FICTURE 使用多层 Dirichlet 模型对像素级空间因子进行随机变量推断,其效率比现有方法高出几个数量级。FICTURE 可以揭示具有挑战性的组织的微观 ST 结构,如真实数据中的血管、纤维化、肌肉和脂质沉积区域,而以往的方法都无法做到这一点。FICTURE 的跨平台通用性、可扩展性和精确性使其成为探索高分辨率 ST 的强大工具。
期刊介绍:
Nature Methods is a monthly journal that focuses on publishing innovative methods and substantial enhancements to fundamental life sciences research techniques. Geared towards a diverse, interdisciplinary readership of researchers in academia and industry engaged in laboratory work, the journal offers new tools for research and emphasizes the immediate practical significance of the featured work. It publishes primary research papers and reviews recent technical and methodological advancements, with a particular interest in primary methods papers relevant to the biological and biomedical sciences. This includes methods rooted in chemistry with practical applications for studying biological problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
