E. David Cohen , Kyle Roethlin , Min Yee , Collynn F. Woeller , Paul S. Brookes , George A. Porter Jr. , Michael A. O'Reilly
{"title":"PPARγ drives mitochondrial stress signaling and the loss of atrial cardiomyocytes in newborn mice exposed to hyperoxia","authors":"E. David Cohen , Kyle Roethlin , Min Yee , Collynn F. Woeller , Paul S. Brookes , George A. Porter Jr. , Michael A. O'Reilly","doi":"10.1016/j.redox.2024.103351","DOIUrl":null,"url":null,"abstract":"<div><p>Diastolic dysfunction is increasingly common in preterm infants exposed to supplemental oxygen (hyperoxia). Previous studies in neonatal mice showed hyperoxia suppresses fatty acid synthesis genes required for proliferation and survival of atrial cardiomyocytes. The loss of atrial cardiomyocytes creates a hypoplastic left atrium that inappropriately fills the left ventricle during diastole. Here, we show that hyperoxia stimulates adenosine monophosphate-activated kinase (AMPK) and peroxisome proliferator activated receptor-gamma (PPARγ) signaling in atrial cardiomyocytes. While both pathways can regulate lipid homeostasis, PPARγ was the primary pathway by which hyperoxia inhibits fatty acid gene expression and inhibits proliferation of mouse atrial HL-1 cells. It also enhanced the toxicity of hyperoxia by increasing expression of activating transcription factor (ATF) 5 and other mitochondrial stress response genes. Silencing PPARγ signaling restored proliferation and survival of HL-1 cells as well as atrial cardiomyocytes in neonatal mice exposed to hyperoxia. Our findings reveal PPARγ enhances the toxicity of hyperoxia on atrial cardiomyocytes, thus suggesting inhibitors of PPARγ signaling may prevent diastolic dysfunction in preterm infants.</p></div>","PeriodicalId":20998,"journal":{"name":"Redox Biology","volume":"76 ","pages":"Article 103351"},"PeriodicalIF":11.9000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S221323172400329X/pdfft?md5=25cda8a9cf5613da51752caefef98c1e&pid=1-s2.0-S221323172400329X-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Redox Biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S221323172400329X","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/12 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
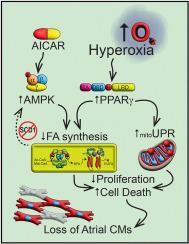
Diastolic dysfunction is increasingly common in preterm infants exposed to supplemental oxygen (hyperoxia). Previous studies in neonatal mice showed hyperoxia suppresses fatty acid synthesis genes required for proliferation and survival of atrial cardiomyocytes. The loss of atrial cardiomyocytes creates a hypoplastic left atrium that inappropriately fills the left ventricle during diastole. Here, we show that hyperoxia stimulates adenosine monophosphate-activated kinase (AMPK) and peroxisome proliferator activated receptor-gamma (PPARγ) signaling in atrial cardiomyocytes. While both pathways can regulate lipid homeostasis, PPARγ was the primary pathway by which hyperoxia inhibits fatty acid gene expression and inhibits proliferation of mouse atrial HL-1 cells. It also enhanced the toxicity of hyperoxia by increasing expression of activating transcription factor (ATF) 5 and other mitochondrial stress response genes. Silencing PPARγ signaling restored proliferation and survival of HL-1 cells as well as atrial cardiomyocytes in neonatal mice exposed to hyperoxia. Our findings reveal PPARγ enhances the toxicity of hyperoxia on atrial cardiomyocytes, thus suggesting inhibitors of PPARγ signaling may prevent diastolic dysfunction in preterm infants.
期刊介绍:
Redox Biology is the official journal of the Society for Redox Biology and Medicine and the Society for Free Radical Research-Europe. It is also affiliated with the International Society for Free Radical Research (SFRRI). This journal serves as a platform for publishing pioneering research, innovative methods, and comprehensive review articles in the field of redox biology, encompassing both health and disease.
Redox Biology welcomes various forms of contributions, including research articles (short or full communications), methods, mini-reviews, and commentaries. Through its diverse range of published content, Redox Biology aims to foster advancements and insights in the understanding of redox biology and its implications.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
