Sarah E. Mudra, Kaleb Ardoin, Vanya Aggarwal, Garrett Diltz, Pedro E. Alcedo Andrade, Catherine M. Broome
{"title":"Thrombotic thrombocytopenic purpura masquerading as Evans syndrome","authors":"Sarah E. Mudra, Kaleb Ardoin, Vanya Aggarwal, Garrett Diltz, Pedro E. Alcedo Andrade, Catherine M. Broome","doi":"10.1002/ajh.27476","DOIUrl":null,"url":null,"abstract":"<p>A 26-year-old female with a history of chronic urticaria (treated with omalizumab in the past) and recently treated chlamydia trachomatis infection presented to an outside emergency department after a syncopal episode. She endorsed a three-day history of nausea, vomiting, fatigue, and ecchymoses. Initial laboratory analysis revealed macrocytic anemia (hemoglobin 4.6 g/dL) with a mean corpuscular volume (MCV) of 100.7 fL, thrombocytopenia (2000 plts/μL), indirect hyperbilirubinemia (indirect bilirubin 3.7 mg/dL), lactate dehydrogenase (LDH) 1245 units/L, absolute reticulocyte count of 0.237 million/μL, haptoglobin 1 mg/dL, international normalized ratio (INR) 1.2 and creatinine 0.75 mg/dL. Peripheral blood smear identified numerous spherocytes and microspherocytes, true thrombocytopenia, and increased reticulocytes without evidence of erythrocyte fragmentation (Figure 1). Direct antiglobulin testing (DAT) was positive for IgG (2+) and C3 (2+) at 37°C, although quantitative analysis was not performed. She received one unit of packed red blood cells and one unit of platelets and was transferred to our institution for further evaluation.</p><p>This patient presented with profound thrombocytopenia and hemolytic anemia as evidenced by elevated LDH, elevated reticulocyte count, decreased haptoglobin, and indirect hyperbilirubinemia. The lack of schistocytes or evidence of erythrocyte fragmentation on peripheral smear as well as positive DAT was consistent with Evans syndrome—warm autoimmune hemolytic anemia (AIHA) with concomitant immune thrombocytopenia (ITP). Evans syndrome can arise spontaneously (primary) or secondary to diseases that generate autoantibodies. Etiologies may include infections, autoimmune disorders, lymphoproliferative disorders, or pregnancy.</p><p>On physical exam, the patient was well-appearing. She was afebrile with the remainder of her vital signs within normal limits. Physical examination was only notable for mild gingival bleeding. No rashes, petechiae, purpura, or ecchymoses were identified. She conversed appropriately and was oriented to person, location, and time. She had not received a blood product transfusion prior to this admission. She did not have a history of pregnancy. Family history was pertinent for Hashimoto's thyroiditis in her mother.</p><p>Overall, the patient appeared clinically well. She reported mild and nonspecific infectious symptoms including malaise, nausea, and emesis after recent domestic travel with her family. No other family members reported similar symptoms. Additionally, she lacked red flag signs and symptoms for malignancy or autoimmune disease including weight loss, night sweats, lymphadenopathy, myalgias, arthralgias, and rash. Nevertheless, broad workup for underlying infectious etiology, autoimmune disease and malignancy was pursued.</p><p>Infectious workup included negative bacterial blood cultures, EBV, CMV, HIV, HSV-1, HSV-2, HHV-6, hepatitis A, B, and C serologies and gastrointestinal pathogen panel. Autoimmune labs were negative for antinuclear antibodies (ANA) as well as antibodies against double-stranded DNA, Sjögren's-syndrome-related antigen A (SSA), Sjögren's-syndrome-related antigen B (SSB), and Smith. Thyroid function tests were within normal limits. Peripheral blood flow cytometry was sent given concern for atypical lymphocytes on further review of peripheral smear as well as hepatomegaly (22 cm in craniocaudal dimension) as noted on computed tomography (CT) abdominal imaging. Flow cytometry and positron emission tomography were both negative for a malignant process.</p><p>The above workup for secondary precipitants of Evans syndrome was unrevealing. Thus, idiopathic AIHA with concomitant ITP was most likely. As such, the patient was started on intravenous immunoglobulin (IVIG, 1 mg/kg) and high dose prednisone (1 mg/kg).</p><p>Her hemoglobin responded appropriately to transfusions, but her platelet count remained low despite high-dose steroids and IVIG. On day three of hospital admission, the patient endorsed acute onset headaches and new right arm weakness. On hospital day four, she was found to be obtunded. Urgent neurologic imaging was obtained with head CT negative for acute hemorrhage. Brain magnetic resonance imaging (MRI) demonstrated cortical hypoxia raising concern for encephalitis or seizures. She was transferred to the medical intensive care unit (MICU) for neurologic monitoring.</p><p>This acute change in clinical status now raised high suspicion for a life-threatening, alternate diagnosis. With new onset neurologic manifestations, thrombotic microangiopathies (TMAs)—most notably, thrombotic thrombocytopenic purpura (TTP)—ascended on the differential as a diagnosis that must be considered. Thus, the medical team reconsidered critical data that argued against MAHA—the positive DAT results and lack of erythrocyte fragmentation on peripheral smear.</p><p>On presentation, the patient lacked hallmark manifestations of TTP including fever and encephalopathy. However, the classic pentad of fever, anemia, thrombocytopenia, renal, and neurologic symptoms is rarely observed.<span><sup>1</sup></span> Patients often present with few of these symptoms or nonspecific symptoms that mimic other etiologies. Furthermore, neurologic manifestations of TTP range from mild confusion to acute ischemic stroke in greater than 60% of patients.<span><sup>2, 3</sup></span> Prompt diagnosis and treatment are tantamount, as without therapy, mortality rates approach 90%.<span><sup>4</sup></span> Even with timely plasma exchange (PLEX) and corticosteroids, mortality remains 10%–15%.<span><sup>4</sup></span></p><p>The PLASMIC score is a validated, clinical diagnostic tool that aids clinicians in assessing the likelihood of severe a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) deficiency.<span><sup>5</sup></span> Notably, the PLASMIC score is only validated in those with TMAs. Thus, although the patient had a PLASMIC score of 6 on admission—placing her in the high-risk category for TTP (with a 72% risk of severe ADAMTS13 deficiency)—her peripheral smear lacked clear evidence of intravascular hemolysis indicative of a MAHA.<span><sup>6</sup></span> Thus, empiric PLEX was deferred. Nevertheless, ADAMTS13 activity level was sent stat on admission; results were pending.</p><p>Additional examination of her peripheral smear on day four of hospitalization demonstrated many nucleated erythrocytes, acanthocytes, and atypical lymphocytes (Figure 2). Still, no schistocytes were noted. ADAMTS13 activity level returned undetectable and identified an ADAMTS13 inhibitor (anti-ADAMTS13 antibodies) at a titer of 2.5. Urgent PLEX was initiated, and high-dose corticosteroids continued. A peripheral blood smear from the subsequent day revealed increased numbers of schistocytes (Figure 3). After two sessions of PLEX, her hemoglobin, platelets, LDH, and total bilirubin all improved. With further PLEX sessions, her hemoglobin reached 10.3 g/dL, platelets reached 248 000 plts/μL, LDH 337 units/L, indirect bilirubin 0.2 mg/dL. Her mental status gradually returned to baseline. Her ADAMTS13 level improved from undetectable to 63% after four sessions of PLEX. Her DAT continued to remain positive for IgG and C3, though titers decreased from 2+ to only weakly positive.</p><p>This case underscores the need for clinicians to consider alternative diagnoses in the face of new data and a rapidly changing clinical course. Persistent anemia and thrombocytopenia despite IVIG and prednisone with the development of new acute encephalopathy demanded an alternate diagnosis. Despite initial data supporting Evans syndrome, TMA—most particularly TTP—became much more likely. When ADAMTS13 activity returned undetectable, TTP was confirmed. PLEX treatment was swiftly initiated.</p><p>Despite continued PLEX, platelets precipitously dropped from 218 000 to 25 000 plts/μL within 3 days. LDH also increased from 365 units/L to 659 units/L. Rituximab 375 mg/m<span><sup>2</sup></span> weekly was added. Daily PLEX sessions continued. Interval ADAMTS13 testing again returned undetectable and identified an increase in ADAMTS13 inhibitor with a titer of 11.1. Thrombocytopenia continued to worsen, reaching a nadir of 14 000 plts/μL at which time caplacizumab was initiated for recalcitrant TTP. After 1 week of caplacizumab, her ADAMTS13 activity increased to 25% with undetectable ADAMTS13 inhibitor and normal hemoglobin and platelet count. A peripheral blood smear showed decreased number of schistocytes (Figure 4). Following 1 month of caplacizumab, ADAMTS13 activity fluctuated to 17%, but counts remained stable. Caplacizumab therapy was extended to 6 weeks and after also completing four weekly doses of rituximab, her ADAMTS13 improved to 21%.</p><p>Broadly, TMAs present with three hallmark features: (1) MAHA, (2) thrombocytopenia, and (3) end-organ damage (Figure 5B). Differential diagnosis for TMA is wide, including primary TMAs and TMAs associated with rheumatologic, neoplastic, renal, and infectious diseases and drug-mediated etiologies (Figure 5A).</p><p>TTP is a rare, life-threatening TMA caused by deficiency of ADAMTS13, a zinc-metalloprotease that cleaves von Willebrand factor (vWF). The estimated incidence is two patients per million per year.<span><sup>7</sup></span> Studies have demonstrated an increased risk in African Americans and females.<span><sup>8</sup></span> To our knowledge, this is the first reported case of TTP masquerading as warm AIHA in an adult which was successfully treated.</p><p>The pathophysiology of TTP involves either acquired autoantibodies impairing ADAMTS13 activity or inherited gene mutations decreasing ADAMTS13 production. ADAMTS13 cleaves vWF. When ADAMTS13 levels are severely reduced (<10%), vWF multimers aggregate, causing resultant microthrombi.<span><sup>8, 9</sup></span> Neurologic and renal impairment result from microthrombi accumulation and subsequent tissue ischemia.<span><sup>3</sup></span> Thrombocytopenia results from platelet consumption and anemia from the hemolysis of red blood cells.<span><sup>3</sup></span></p><p>Management of TTP requires prompt treatment initiation. First-line therapy involves PLEX and steroids. PLEX removes circulating anti-ADAMTS13 autoantibodies and restores ADAMTS13 function.<span><sup>10</sup></span> Immunosuppression via corticosteroids dampens the autoimmune response perpetuating microthrombi generation.<span><sup>11</sup></span> Additional agents include rituximab, a monoclonal antibody against CD20, and caplacizumab, a humanized bivalent immunoglobulin fragment targeting the A1 domain of vWF.<span><sup>12</sup></span> Rituximab may be added in the acute phase to aid in achieving relapse and for patients with refractory disease.<span><sup>8, 11</sup></span> Caplacizumab prevents interaction with the platelet glycoprotein Ib-IX-V receptor and thereby decreases microvascular hemolysis. Caplacizumab has been shown to decrease rates of TTP recurrence, TTP-related death, and yield faster resolution of thrombocytopenia.<span><sup>7</sup></span></p><p>Uniquely, this case posed a diagnostic challenge when the DAT was positive for both IgG and C3. Classically, TMAs like TTP present with a DAT negative hemolytic anemia while autoimmune and drug-induced hemolytic anemias result in a positive DAT.<span><sup>13</sup></span> A positive DAT supported a diagnosis of Evans syndrome (warm AIHA and ITP). Furthermore, review of multiple peripheral smears from the first few days of admission lacked schistocytes. Instead, spherocytes and microspherocytes were observed, suggesting extravascular rather than intravascular hemolysis. It has been reported that microspherocytes can be a feature of TMA-related MAHA.</p><p>Prior administration of IVIG or red blood cell transfusions may also result in positive DATs. Notably, the DAT was performed in this case prior to blood transfusion and IVIG administration. Rare cases of TTP with positive DATs have been reported among those with autoimmune diseases including systemic lupus erythematosus (SLE) and systemic connective tissue diseases.<span><sup>14-17</sup></span> In one case series of six patients with positive DATs and diagnosed TTP, 50% had concomitant SLE.<span><sup>15</sup></span> There are three previously reported cases of TTP that were initially diagnosed as Evans syndrome based on a positive DAT, two in adults<span><sup>18, 19</sup></span> and one in a child.<span><sup>13</sup></span> The previously reported adult cases resulted in death before initiation of PLEX<span><sup>18, 19</sup></span> and in the case reported by Zhang, Chen, and Zhang,<span><sup>19</sup></span> the peripheral smear did not show schistocytes until the third day of admission when renal function declined—which then prompted concern for TTP.</p><p>Certainly, further investigation is needed to clarify the mechanism of DAT positivity in TTP—especially when positive DAT testing classically points away from TTP and even urgent ADAMTS13 assays may take days to result. It is possible that the patient's hemolysis was being driven by both the warm autoantibodies and TTP, for which rituximab should target both pathways. Further education as to the myriad of peripheral blood smear findings in MAHA is needed to allow for a wide differential diagnosis even when classic schistocytes are not present.</p><p>In summary, TTP represents a rare, life-threatening TMA requiring prompt initiation of urgent PLEX and corticosteroids. Physicians must remain vigilant given the high morbidity and mortality associated with TTP. Additionally, this case underscores its variable and nonspecific presentation that mimicked Evans syndrome—a disease with a more benign course. TTP should remain in the differential diagnosis when a hemolytic anemia accompanied by thrombocytopenia fails to improve with standard treatment. This case highlights the urgent need for quicker turnaround time of ADAMTS13 testing.</p><p>No funding was used in the preparation of this article.</p><p>The authors declare no conflicts of interest.</p><p>Written consent was obtained from the patient.</p>","PeriodicalId":7724,"journal":{"name":"American Journal of Hematology","volume":"99 12","pages":"2381-2385"},"PeriodicalIF":9.9000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajh.27476","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Hematology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ajh.27476","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
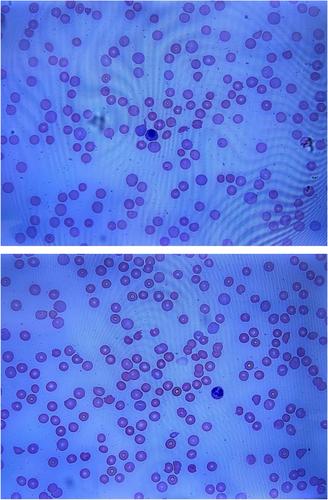
A 26-year-old female with a history of chronic urticaria (treated with omalizumab in the past) and recently treated chlamydia trachomatis infection presented to an outside emergency department after a syncopal episode. She endorsed a three-day history of nausea, vomiting, fatigue, and ecchymoses. Initial laboratory analysis revealed macrocytic anemia (hemoglobin 4.6 g/dL) with a mean corpuscular volume (MCV) of 100.7 fL, thrombocytopenia (2000 plts/μL), indirect hyperbilirubinemia (indirect bilirubin 3.7 mg/dL), lactate dehydrogenase (LDH) 1245 units/L, absolute reticulocyte count of 0.237 million/μL, haptoglobin 1 mg/dL, international normalized ratio (INR) 1.2 and creatinine 0.75 mg/dL. Peripheral blood smear identified numerous spherocytes and microspherocytes, true thrombocytopenia, and increased reticulocytes without evidence of erythrocyte fragmentation (Figure 1). Direct antiglobulin testing (DAT) was positive for IgG (2+) and C3 (2+) at 37°C, although quantitative analysis was not performed. She received one unit of packed red blood cells and one unit of platelets and was transferred to our institution for further evaluation.
This patient presented with profound thrombocytopenia and hemolytic anemia as evidenced by elevated LDH, elevated reticulocyte count, decreased haptoglobin, and indirect hyperbilirubinemia. The lack of schistocytes or evidence of erythrocyte fragmentation on peripheral smear as well as positive DAT was consistent with Evans syndrome—warm autoimmune hemolytic anemia (AIHA) with concomitant immune thrombocytopenia (ITP). Evans syndrome can arise spontaneously (primary) or secondary to diseases that generate autoantibodies. Etiologies may include infections, autoimmune disorders, lymphoproliferative disorders, or pregnancy.
On physical exam, the patient was well-appearing. She was afebrile with the remainder of her vital signs within normal limits. Physical examination was only notable for mild gingival bleeding. No rashes, petechiae, purpura, or ecchymoses were identified. She conversed appropriately and was oriented to person, location, and time. She had not received a blood product transfusion prior to this admission. She did not have a history of pregnancy. Family history was pertinent for Hashimoto's thyroiditis in her mother.
Overall, the patient appeared clinically well. She reported mild and nonspecific infectious symptoms including malaise, nausea, and emesis after recent domestic travel with her family. No other family members reported similar symptoms. Additionally, she lacked red flag signs and symptoms for malignancy or autoimmune disease including weight loss, night sweats, lymphadenopathy, myalgias, arthralgias, and rash. Nevertheless, broad workup for underlying infectious etiology, autoimmune disease and malignancy was pursued.
Infectious workup included negative bacterial blood cultures, EBV, CMV, HIV, HSV-1, HSV-2, HHV-6, hepatitis A, B, and C serologies and gastrointestinal pathogen panel. Autoimmune labs were negative for antinuclear antibodies (ANA) as well as antibodies against double-stranded DNA, Sjögren's-syndrome-related antigen A (SSA), Sjögren's-syndrome-related antigen B (SSB), and Smith. Thyroid function tests were within normal limits. Peripheral blood flow cytometry was sent given concern for atypical lymphocytes on further review of peripheral smear as well as hepatomegaly (22 cm in craniocaudal dimension) as noted on computed tomography (CT) abdominal imaging. Flow cytometry and positron emission tomography were both negative for a malignant process.
The above workup for secondary precipitants of Evans syndrome was unrevealing. Thus, idiopathic AIHA with concomitant ITP was most likely. As such, the patient was started on intravenous immunoglobulin (IVIG, 1 mg/kg) and high dose prednisone (1 mg/kg).
Her hemoglobin responded appropriately to transfusions, but her platelet count remained low despite high-dose steroids and IVIG. On day three of hospital admission, the patient endorsed acute onset headaches and new right arm weakness. On hospital day four, she was found to be obtunded. Urgent neurologic imaging was obtained with head CT negative for acute hemorrhage. Brain magnetic resonance imaging (MRI) demonstrated cortical hypoxia raising concern for encephalitis or seizures. She was transferred to the medical intensive care unit (MICU) for neurologic monitoring.
This acute change in clinical status now raised high suspicion for a life-threatening, alternate diagnosis. With new onset neurologic manifestations, thrombotic microangiopathies (TMAs)—most notably, thrombotic thrombocytopenic purpura (TTP)—ascended on the differential as a diagnosis that must be considered. Thus, the medical team reconsidered critical data that argued against MAHA—the positive DAT results and lack of erythrocyte fragmentation on peripheral smear.
On presentation, the patient lacked hallmark manifestations of TTP including fever and encephalopathy. However, the classic pentad of fever, anemia, thrombocytopenia, renal, and neurologic symptoms is rarely observed.1 Patients often present with few of these symptoms or nonspecific symptoms that mimic other etiologies. Furthermore, neurologic manifestations of TTP range from mild confusion to acute ischemic stroke in greater than 60% of patients.2, 3 Prompt diagnosis and treatment are tantamount, as without therapy, mortality rates approach 90%.4 Even with timely plasma exchange (PLEX) and corticosteroids, mortality remains 10%–15%.4
The PLASMIC score is a validated, clinical diagnostic tool that aids clinicians in assessing the likelihood of severe a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) deficiency.5 Notably, the PLASMIC score is only validated in those with TMAs. Thus, although the patient had a PLASMIC score of 6 on admission—placing her in the high-risk category for TTP (with a 72% risk of severe ADAMTS13 deficiency)—her peripheral smear lacked clear evidence of intravascular hemolysis indicative of a MAHA.6 Thus, empiric PLEX was deferred. Nevertheless, ADAMTS13 activity level was sent stat on admission; results were pending.
Additional examination of her peripheral smear on day four of hospitalization demonstrated many nucleated erythrocytes, acanthocytes, and atypical lymphocytes (Figure 2). Still, no schistocytes were noted. ADAMTS13 activity level returned undetectable and identified an ADAMTS13 inhibitor (anti-ADAMTS13 antibodies) at a titer of 2.5. Urgent PLEX was initiated, and high-dose corticosteroids continued. A peripheral blood smear from the subsequent day revealed increased numbers of schistocytes (Figure 3). After two sessions of PLEX, her hemoglobin, platelets, LDH, and total bilirubin all improved. With further PLEX sessions, her hemoglobin reached 10.3 g/dL, platelets reached 248 000 plts/μL, LDH 337 units/L, indirect bilirubin 0.2 mg/dL. Her mental status gradually returned to baseline. Her ADAMTS13 level improved from undetectable to 63% after four sessions of PLEX. Her DAT continued to remain positive for IgG and C3, though titers decreased from 2+ to only weakly positive.
This case underscores the need for clinicians to consider alternative diagnoses in the face of new data and a rapidly changing clinical course. Persistent anemia and thrombocytopenia despite IVIG and prednisone with the development of new acute encephalopathy demanded an alternate diagnosis. Despite initial data supporting Evans syndrome, TMA—most particularly TTP—became much more likely. When ADAMTS13 activity returned undetectable, TTP was confirmed. PLEX treatment was swiftly initiated.
Despite continued PLEX, platelets precipitously dropped from 218 000 to 25 000 plts/μL within 3 days. LDH also increased from 365 units/L to 659 units/L. Rituximab 375 mg/m2 weekly was added. Daily PLEX sessions continued. Interval ADAMTS13 testing again returned undetectable and identified an increase in ADAMTS13 inhibitor with a titer of 11.1. Thrombocytopenia continued to worsen, reaching a nadir of 14 000 plts/μL at which time caplacizumab was initiated for recalcitrant TTP. After 1 week of caplacizumab, her ADAMTS13 activity increased to 25% with undetectable ADAMTS13 inhibitor and normal hemoglobin and platelet count. A peripheral blood smear showed decreased number of schistocytes (Figure 4). Following 1 month of caplacizumab, ADAMTS13 activity fluctuated to 17%, but counts remained stable. Caplacizumab therapy was extended to 6 weeks and after also completing four weekly doses of rituximab, her ADAMTS13 improved to 21%.
Broadly, TMAs present with three hallmark features: (1) MAHA, (2) thrombocytopenia, and (3) end-organ damage (Figure 5B). Differential diagnosis for TMA is wide, including primary TMAs and TMAs associated with rheumatologic, neoplastic, renal, and infectious diseases and drug-mediated etiologies (Figure 5A).
TTP is a rare, life-threatening TMA caused by deficiency of ADAMTS13, a zinc-metalloprotease that cleaves von Willebrand factor (vWF). The estimated incidence is two patients per million per year.7 Studies have demonstrated an increased risk in African Americans and females.8 To our knowledge, this is the first reported case of TTP masquerading as warm AIHA in an adult which was successfully treated.
The pathophysiology of TTP involves either acquired autoantibodies impairing ADAMTS13 activity or inherited gene mutations decreasing ADAMTS13 production. ADAMTS13 cleaves vWF. When ADAMTS13 levels are severely reduced (<10%), vWF multimers aggregate, causing resultant microthrombi.8, 9 Neurologic and renal impairment result from microthrombi accumulation and subsequent tissue ischemia.3 Thrombocytopenia results from platelet consumption and anemia from the hemolysis of red blood cells.3
Management of TTP requires prompt treatment initiation. First-line therapy involves PLEX and steroids. PLEX removes circulating anti-ADAMTS13 autoantibodies and restores ADAMTS13 function.10 Immunosuppression via corticosteroids dampens the autoimmune response perpetuating microthrombi generation.11 Additional agents include rituximab, a monoclonal antibody against CD20, and caplacizumab, a humanized bivalent immunoglobulin fragment targeting the A1 domain of vWF.12 Rituximab may be added in the acute phase to aid in achieving relapse and for patients with refractory disease.8, 11 Caplacizumab prevents interaction with the platelet glycoprotein Ib-IX-V receptor and thereby decreases microvascular hemolysis. Caplacizumab has been shown to decrease rates of TTP recurrence, TTP-related death, and yield faster resolution of thrombocytopenia.7
Uniquely, this case posed a diagnostic challenge when the DAT was positive for both IgG and C3. Classically, TMAs like TTP present with a DAT negative hemolytic anemia while autoimmune and drug-induced hemolytic anemias result in a positive DAT.13 A positive DAT supported a diagnosis of Evans syndrome (warm AIHA and ITP). Furthermore, review of multiple peripheral smears from the first few days of admission lacked schistocytes. Instead, spherocytes and microspherocytes were observed, suggesting extravascular rather than intravascular hemolysis. It has been reported that microspherocytes can be a feature of TMA-related MAHA.
Prior administration of IVIG or red blood cell transfusions may also result in positive DATs. Notably, the DAT was performed in this case prior to blood transfusion and IVIG administration. Rare cases of TTP with positive DATs have been reported among those with autoimmune diseases including systemic lupus erythematosus (SLE) and systemic connective tissue diseases.14-17 In one case series of six patients with positive DATs and diagnosed TTP, 50% had concomitant SLE.15 There are three previously reported cases of TTP that were initially diagnosed as Evans syndrome based on a positive DAT, two in adults18, 19 and one in a child.13 The previously reported adult cases resulted in death before initiation of PLEX18, 19 and in the case reported by Zhang, Chen, and Zhang,19 the peripheral smear did not show schistocytes until the third day of admission when renal function declined—which then prompted concern for TTP.
Certainly, further investigation is needed to clarify the mechanism of DAT positivity in TTP—especially when positive DAT testing classically points away from TTP and even urgent ADAMTS13 assays may take days to result. It is possible that the patient's hemolysis was being driven by both the warm autoantibodies and TTP, for which rituximab should target both pathways. Further education as to the myriad of peripheral blood smear findings in MAHA is needed to allow for a wide differential diagnosis even when classic schistocytes are not present.
In summary, TTP represents a rare, life-threatening TMA requiring prompt initiation of urgent PLEX and corticosteroids. Physicians must remain vigilant given the high morbidity and mortality associated with TTP. Additionally, this case underscores its variable and nonspecific presentation that mimicked Evans syndrome—a disease with a more benign course. TTP should remain in the differential diagnosis when a hemolytic anemia accompanied by thrombocytopenia fails to improve with standard treatment. This case highlights the urgent need for quicker turnaround time of ADAMTS13 testing.
No funding was used in the preparation of this article.
期刊介绍:
The American Journal of Hematology offers extensive coverage of experimental and clinical aspects of blood diseases in humans and animal models. The journal publishes original contributions in both non-malignant and malignant hematological diseases, encompassing clinical and basic studies in areas such as hemostasis, thrombosis, immunology, blood banking, and stem cell biology. Clinical translational reports highlighting innovative therapeutic approaches for the diagnosis and treatment of hematological diseases are actively encouraged.The American Journal of Hematology features regular original laboratory and clinical research articles, brief research reports, critical reviews, images in hematology, as well as letters and correspondence.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
