Yingying Jiang , Xiaogang Luo , Zhanpeng Zheng , Shun Wen , Hongwei Gao , Cheng Xu , Min Jiang , Siyuan Wang
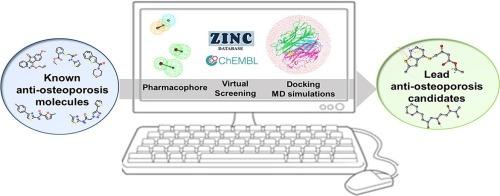
{"title":"Identification of novel RANKL inhibitors through in silico analysis","authors":"Yingying Jiang , Xiaogang Luo , Zhanpeng Zheng , Shun Wen , Hongwei Gao , Cheng Xu , Min Jiang , Siyuan Wang","doi":"10.1016/j.bioorg.2024.107826","DOIUrl":null,"url":null,"abstract":"<div><p>Receptor activator of nuclear factor-κB ligand (RANKL) is considered the principal regulator of osteoclast differentiation. Therefore, strategies interfering with the RANKL-RANK signaling pathway may effectively inhibit osteoclast differentiation and mitigate bone resorption. Consequently, RANKL has become a promising target for new drug design strategies. Despite extensive research on specific drugs and antibodies, only a few have shown efficacy in treating osteoporosis. To address this challenge, we aimed to explore new approaches for designing drugs for osteoporosis. In this study, a 3D quantitative structure–activity relationship (QSAR) pharmacophore model was built for RANKL with reference to known inhibitor IC<sub>50</sub> values. The optimal pharmacophore model was then employed as a 3D query to screen databases for novel lead compounds. The obtained compounds were subjected to ADMET and TOPKAT analyses to predict drug pharmacokinetics and toxicity. Molecular docking and <em>de novo</em> evolution approaches were applied to verify the docking binding affinities of the compounds. Five candidate compounds were subjected to further <em>in vitro</em> analyses to assess their anti-osteoporotic effects, among which compound <strong>4</strong> demonstrated significant inhibitory activity, achieving an inhibitory rate of 92.6 % on osteoclastogenesis at a concentration of 10 μM. Subsequent molecular dynamics (MD) simulations to assess the stability and behavior of compound <strong>4</strong> and its evolved variant, ZINC00059014397_Evo, within the RANKL binding site revealed that the variant is a potential therapeutic agent for targeting osteoclasts. This study offers valuable insights for developing next generation RANKL inhibitors for osteoporosis treatments.</p></div>","PeriodicalId":257,"journal":{"name":"Bioorganic Chemistry","volume":"153 ","pages":"Article 107826"},"PeriodicalIF":4.7000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioorganic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0045206824007314","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/16 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Receptor activator of nuclear factor-κB ligand (RANKL) is considered the principal regulator of osteoclast differentiation. Therefore, strategies interfering with the RANKL-RANK signaling pathway may effectively inhibit osteoclast differentiation and mitigate bone resorption. Consequently, RANKL has become a promising target for new drug design strategies. Despite extensive research on specific drugs and antibodies, only a few have shown efficacy in treating osteoporosis. To address this challenge, we aimed to explore new approaches for designing drugs for osteoporosis. In this study, a 3D quantitative structure–activity relationship (QSAR) pharmacophore model was built for RANKL with reference to known inhibitor IC50 values. The optimal pharmacophore model was then employed as a 3D query to screen databases for novel lead compounds. The obtained compounds were subjected to ADMET and TOPKAT analyses to predict drug pharmacokinetics and toxicity. Molecular docking and de novo evolution approaches were applied to verify the docking binding affinities of the compounds. Five candidate compounds were subjected to further in vitro analyses to assess their anti-osteoporotic effects, among which compound 4 demonstrated significant inhibitory activity, achieving an inhibitory rate of 92.6 % on osteoclastogenesis at a concentration of 10 μM. Subsequent molecular dynamics (MD) simulations to assess the stability and behavior of compound 4 and its evolved variant, ZINC00059014397_Evo, within the RANKL binding site revealed that the variant is a potential therapeutic agent for targeting osteoclasts. This study offers valuable insights for developing next generation RANKL inhibitors for osteoporosis treatments.
期刊介绍:
Bioorganic Chemistry publishes research that addresses biological questions at the molecular level, using organic chemistry and principles of physical organic chemistry. The scope of the journal covers a range of topics at the organic chemistry-biology interface, including: enzyme catalysis, biotransformation and enzyme inhibition; nucleic acids chemistry; medicinal chemistry; natural product chemistry, natural product synthesis and natural product biosynthesis; antimicrobial agents; lipid and peptide chemistry; biophysical chemistry; biological probes; bio-orthogonal chemistry and biomimetic chemistry.
For manuscripts dealing with synthetic bioactive compounds, the Journal requires that the molecular target of the compounds described must be known, and must be demonstrated experimentally in the manuscript. For studies involving natural products, if the molecular target is unknown, some data beyond simple cell-based toxicity studies to provide insight into the mechanism of action is required. Studies supported by molecular docking are welcome, but must be supported by experimental data. The Journal does not consider manuscripts that are purely theoretical or computational in nature.
The Journal publishes regular articles, short communications and reviews. Reviews are normally invited by Editors or Editorial Board members. Authors of unsolicited reviews should first contact an Editor or Editorial Board member to determine whether the proposed article is within the scope of the Journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
