Bidyadhar Sethy , Zih-Yao Yu , Iin Narwanti , Richa Upadhyay , Mei-Jung Lai , Sung-Bau Lee , Jing-Ping Liou
{"title":"Design, synthesis, and biological evaluation of adenosine derivatives targeting DOT1L and HAT as anti-leukemia agents","authors":"Bidyadhar Sethy , Zih-Yao Yu , Iin Narwanti , Richa Upadhyay , Mei-Jung Lai , Sung-Bau Lee , Jing-Ping Liou","doi":"10.1016/j.bioorg.2024.107771","DOIUrl":null,"url":null,"abstract":"<div><p>Disruptor of telomeric silencing 1-like (DOT1L) is a key hub in histone lysine methyltransferase and an attractive therapeutic target for treating hematological malignancies including acute myeloid leukemia (AML). In this study, we report the design and synthesis of a new series of adenosine derivatives as DOT1L inhibitors by accommodating a basic linker piperidine-4-ylmethyl motif to respective aryl-urea/benzimidazole scaffolds. The anti-DOT1L enzyme activity analysis demonstrated that compounds <strong>8</strong>, <strong>12,</strong> and <strong>13</strong> strongly suppressed DOT1L activity with IC<sub>50</sub> values ranging from 0.125 to 0.408 µM among all the synthetics, and the structure–activity relationships were summarized. Moreover, compound <strong>12</strong> possessed relatively potent DOT1L inhibitory activity by significantly reduced histone H3 di-methylation at lysine 79 (H3K79me2) level in cells. Subsequently, all the synthetics were screened against various leukemia cell lines, indicating the DOT1L active adenosine derivatives exhibited low to moderate while compound <strong>15</strong> showed strong cellular inhibition despite its unsuccessful DOT1L inhibition. Therefore, acknowledging the distinctive potency of compound <strong>15</strong> against five different leukemia cell lines, including <em>MLL-r</em> (MV4-11) and <em>non-MLL-r</em> cell lines (HL-60, HH, K562, and KG-1), with IC<sub>50</sub> values in the 0.45 ∼ 1.66 μM range and its mode of action was explored. Furthermore, compound <strong>15</strong> hindered histone acetylation, induced remarkable DNA damage, and triggered apoptosis. Importantly, normal T lymphocytes only showed moderate response to compound <strong>15</strong>. These findings provide a basis for future studies on its potential application against AML.</p></div>","PeriodicalId":257,"journal":{"name":"Bioorganic Chemistry","volume":"153 ","pages":"Article 107771"},"PeriodicalIF":4.7000,"publicationDate":"2024-09-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioorganic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S004520682400676X","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
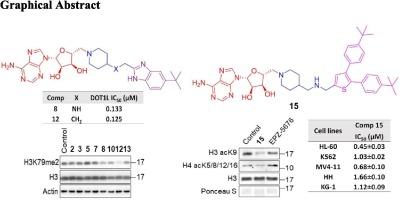
Disruptor of telomeric silencing 1-like (DOT1L) is a key hub in histone lysine methyltransferase and an attractive therapeutic target for treating hematological malignancies including acute myeloid leukemia (AML). In this study, we report the design and synthesis of a new series of adenosine derivatives as DOT1L inhibitors by accommodating a basic linker piperidine-4-ylmethyl motif to respective aryl-urea/benzimidazole scaffolds. The anti-DOT1L enzyme activity analysis demonstrated that compounds 8, 12, and 13 strongly suppressed DOT1L activity with IC50 values ranging from 0.125 to 0.408 µM among all the synthetics, and the structure–activity relationships were summarized. Moreover, compound 12 possessed relatively potent DOT1L inhibitory activity by significantly reduced histone H3 di-methylation at lysine 79 (H3K79me2) level in cells. Subsequently, all the synthetics were screened against various leukemia cell lines, indicating the DOT1L active adenosine derivatives exhibited low to moderate while compound 15 showed strong cellular inhibition despite its unsuccessful DOT1L inhibition. Therefore, acknowledging the distinctive potency of compound 15 against five different leukemia cell lines, including MLL-r (MV4-11) and non-MLL-r cell lines (HL-60, HH, K562, and KG-1), with IC50 values in the 0.45 ∼ 1.66 μM range and its mode of action was explored. Furthermore, compound 15 hindered histone acetylation, induced remarkable DNA damage, and triggered apoptosis. Importantly, normal T lymphocytes only showed moderate response to compound 15. These findings provide a basis for future studies on its potential application against AML.
期刊介绍:
Bioorganic Chemistry publishes research that addresses biological questions at the molecular level, using organic chemistry and principles of physical organic chemistry. The scope of the journal covers a range of topics at the organic chemistry-biology interface, including: enzyme catalysis, biotransformation and enzyme inhibition; nucleic acids chemistry; medicinal chemistry; natural product chemistry, natural product synthesis and natural product biosynthesis; antimicrobial agents; lipid and peptide chemistry; biophysical chemistry; biological probes; bio-orthogonal chemistry and biomimetic chemistry.
For manuscripts dealing with synthetic bioactive compounds, the Journal requires that the molecular target of the compounds described must be known, and must be demonstrated experimentally in the manuscript. For studies involving natural products, if the molecular target is unknown, some data beyond simple cell-based toxicity studies to provide insight into the mechanism of action is required. Studies supported by molecular docking are welcome, but must be supported by experimental data. The Journal does not consider manuscripts that are purely theoretical or computational in nature.
The Journal publishes regular articles, short communications and reviews. Reviews are normally invited by Editors or Editorial Board members. Authors of unsolicited reviews should first contact an Editor or Editorial Board member to determine whether the proposed article is within the scope of the Journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
