{"title":"Density functional theory and molecular dynamics simulation of water molecules confined between two-dimensional graphene oxide surfaces","authors":"Mohsen Abbaspour , Ali Morsali","doi":"10.1016/j.jmgm.2024.108862","DOIUrl":null,"url":null,"abstract":"<div><p>In this work, the interaction potentials of water molecule with the two-dimensional graphene oxide (GO) surfaces containing epoxy groups have been determined using the M06–2X/6-31g (d,p) level of theory at different orientations and separations and fitted to the Born-Huggins-Meyer (BHM) potential. Good agreements were found between the computed and the well-known OPLS-AA and Dreiding potentials. We have also used some calculated potentials and the well-known models in the molecular dynamics (MD) simulations. Our results showed that some of the calculated force fields for both 2D GO structures almost represent similar results of average number of hydrogen bonds (<HB>), radial distribution functions (RDF), self-diffusion coefficient, and angle distribution function (ADF) with the OPLS-AA and Dreiding models which are due to their agreements of the interaction potentials. However, some models in both GO systems represent different results because of their shifted potentials to the larger distances. Our results also showed that the confined water molecules tend to orient toward the epoxy groups on the GO surfaces and the distributions at the angles of θ = 0<sup>o</sup> (or θ = 180<sup>o</sup>) is more than the other distributions. The water molecules confined between the bent GO surfaces showed less diffusion coefficients than the flat structure.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"133 ","pages":"Article 108862"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001621","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/12 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
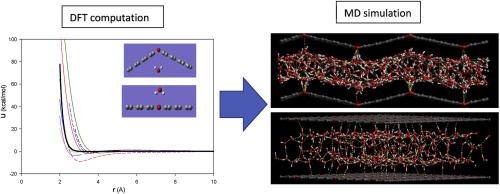
In this work, the interaction potentials of water molecule with the two-dimensional graphene oxide (GO) surfaces containing epoxy groups have been determined using the M06–2X/6-31g (d,p) level of theory at different orientations and separations and fitted to the Born-Huggins-Meyer (BHM) potential. Good agreements were found between the computed and the well-known OPLS-AA and Dreiding potentials. We have also used some calculated potentials and the well-known models in the molecular dynamics (MD) simulations. Our results showed that some of the calculated force fields for both 2D GO structures almost represent similar results of average number of hydrogen bonds (<HB>), radial distribution functions (RDF), self-diffusion coefficient, and angle distribution function (ADF) with the OPLS-AA and Dreiding models which are due to their agreements of the interaction potentials. However, some models in both GO systems represent different results because of their shifted potentials to the larger distances. Our results also showed that the confined water molecules tend to orient toward the epoxy groups on the GO surfaces and the distributions at the angles of θ = 0o (or θ = 180o) is more than the other distributions. The water molecules confined between the bent GO surfaces showed less diffusion coefficients than the flat structure.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
