{"title":"Elucidating the Hydrolysis and Polymerization Reactions of Al3+-Solvated Molecules by Reactive Molecular Dynamics Simulation","authors":"Feng Liu, Qi Zhao, Yuguo Xia, Xiuling Jiao, Dairong Chen","doi":"10.1002/qua.27483","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Utilizing the reactive molecular dynamics (ReaxFF MD) simulation, we conducted a comprehensive study on the impact of basicity (OH<sup>−</sup>/Al<sup>3+</sup> ratio), concentration, and temperature on the hydrolysis and polymerization reactions of Al<sup>3+</sup>-solvated molecules. Through simulations, we analyzed the structural changes, energy fluctuations of the system, and the evolution patterns of reaction products under different parameters, which were subsequently validated by experimental data. The research results indicate that hydroxide ions in the solution directly influence the breakage of O<span></span>H bonds in the coordinating water molecules of solvated aluminum ions. This, in turn, affects the number of H<sub>2</sub>O and OH<sup>−</sup> ions coordinated with Al<sup>3+</sup>, leading to changes in hydrolysis products. Additionally, the number of OH<sup>−</sup> ions surrounding Al<sup>3+</sup> affects the electrostatic repulsion, making it easier for polymerization reactions to occur as the system approaches the point of zero charge. On the other hand, an increase in concentration and temperature enhances the frequency of cluster collisions, thus contributing to an increase in polymerization degree. The experimental results align closely with our simulated predictions. As the pH value increases, the particle size exhibits a trend of first increasing and then decreasing, reaching a maximum at the point of zero charge. Simultaneously, an increase in concentration also prompts an increase in particle size. The combination of these empirical results with simulations enhances the credibility and reliability of our model's predictive capabilities. This study not only expands our understanding of the relevant chemical reaction processes but also provides important theoretical support for practical applications in related fields.</p>\n </div>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 19","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27483","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
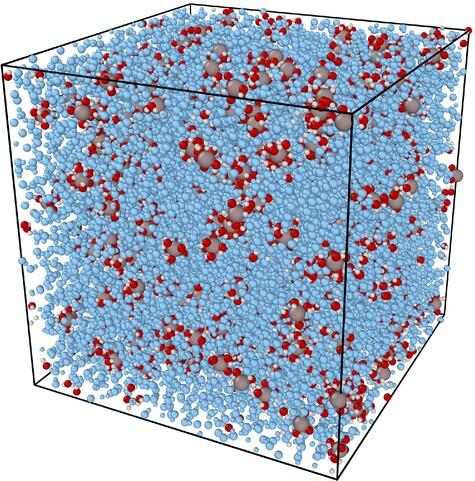
Utilizing the reactive molecular dynamics (ReaxFF MD) simulation, we conducted a comprehensive study on the impact of basicity (OH−/Al3+ ratio), concentration, and temperature on the hydrolysis and polymerization reactions of Al3+-solvated molecules. Through simulations, we analyzed the structural changes, energy fluctuations of the system, and the evolution patterns of reaction products under different parameters, which were subsequently validated by experimental data. The research results indicate that hydroxide ions in the solution directly influence the breakage of OH bonds in the coordinating water molecules of solvated aluminum ions. This, in turn, affects the number of H2O and OH− ions coordinated with Al3+, leading to changes in hydrolysis products. Additionally, the number of OH− ions surrounding Al3+ affects the electrostatic repulsion, making it easier for polymerization reactions to occur as the system approaches the point of zero charge. On the other hand, an increase in concentration and temperature enhances the frequency of cluster collisions, thus contributing to an increase in polymerization degree. The experimental results align closely with our simulated predictions. As the pH value increases, the particle size exhibits a trend of first increasing and then decreasing, reaching a maximum at the point of zero charge. Simultaneously, an increase in concentration also prompts an increase in particle size. The combination of these empirical results with simulations enhances the credibility and reliability of our model's predictive capabilities. This study not only expands our understanding of the relevant chemical reaction processes but also provides important theoretical support for practical applications in related fields.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
