Francesco Berluti, Fady Baselious, Sven Hagemann, Sebastian Hilscher, Matthias Schmidt, Stefan Hüttelmaier, Mike Schutkowski, Wolfgang Sippl, Hany S. Ibrahim
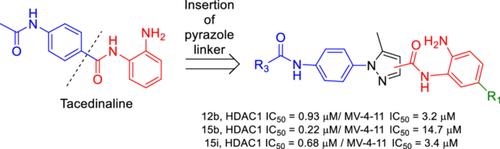
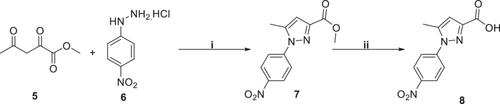
{"title":"Development of new pyrazoles as class I HDAC inhibitors: Synthesis, molecular modeling, and biological characterization in leukemia cells","authors":"Francesco Berluti, Fady Baselious, Sven Hagemann, Sebastian Hilscher, Matthias Schmidt, Stefan Hüttelmaier, Mike Schutkowski, Wolfgang Sippl, Hany S. Ibrahim","doi":"10.1002/ardp.202400437","DOIUrl":null,"url":null,"abstract":"<p>Class I histone deacetylases (HDACs) are considered promising targets in current cancer research. To obtain subtype-selective and potent HDAC inhibitors, we used the aminobenzamide scaffold as the zinc-binding group and prepared new derivatives with a pyrazole ring as the linking group. The synthesized compounds were analyzed in vitro using an enzymatic assay against HDAC1, −2, and −3. Compounds <b>12b</b>, <b>15b</b>, and <b>15i</b> were found to be potent HDAC1 inhibitors, also in comparison to the reference compounds entinostat and tacedinaline, with IC<sub>50</sub> values of 0.93, 0.22, and 0.68 μM, respectively. The best compounds were measured for their cellular effect and target engagement in acute myeloid leukemia (AML) cells. In addition, we studied the interaction of the compounds with HDAC subtypes using docking and molecular dynamic simulations. In summary, we have developed a new chemotype of HDAC1 inhibitors that can be used for further structure-based optimization.</p>","PeriodicalId":128,"journal":{"name":"Archiv der Pharmazie","volume":"357 11","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ardp.202400437","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archiv der Pharmazie","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ardp.202400437","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
Class I histone deacetylases (HDACs) are considered promising targets in current cancer research. To obtain subtype-selective and potent HDAC inhibitors, we used the aminobenzamide scaffold as the zinc-binding group and prepared new derivatives with a pyrazole ring as the linking group. The synthesized compounds were analyzed in vitro using an enzymatic assay against HDAC1, −2, and −3. Compounds 12b, 15b, and 15i were found to be potent HDAC1 inhibitors, also in comparison to the reference compounds entinostat and tacedinaline, with IC50 values of 0.93, 0.22, and 0.68 μM, respectively. The best compounds were measured for their cellular effect and target engagement in acute myeloid leukemia (AML) cells. In addition, we studied the interaction of the compounds with HDAC subtypes using docking and molecular dynamic simulations. In summary, we have developed a new chemotype of HDAC1 inhibitors that can be used for further structure-based optimization.
期刊介绍:
Archiv der Pharmazie - Chemistry in Life Sciences is an international journal devoted to research and development in all fields of pharmaceutical and medicinal chemistry. Emphasis is put on papers combining synthetic organic chemistry, structural biology, molecular modelling, bioorganic chemistry, natural products chemistry, biochemistry or analytical methods with pharmaceutical or medicinal aspects such as biological activity. The focus of this journal is put on original research papers, but other scientifically valuable contributions (e.g. reviews, minireviews, highlights, symposia contributions, discussions, and essays) are also welcome.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
