Martina De Riggi, Agnese De Giorgi, Luca Pollini, Luca Angelini, Giulia Paparella, Antonio Cannavacciuolo, Daniele Birreci, Davide Costa, Alessandra Tessa, Gemma Natale, Marco Fiorelli, Daniele Galatolo, Filippo Maria Santorelli, Serena Galosi, Matteo Bologna
{"title":"CACNA1G Causes Dominantly Inherited Myoclonus-Ataxia with Intellectual Disability: A Case Report","authors":"Martina De Riggi, Agnese De Giorgi, Luca Pollini, Luca Angelini, Giulia Paparella, Antonio Cannavacciuolo, Daniele Birreci, Davide Costa, Alessandra Tessa, Gemma Natale, Marco Fiorelli, Daniele Galatolo, Filippo Maria Santorelli, Serena Galosi, Matteo Bologna","doi":"10.1007/s12311-024-01734-6","DOIUrl":null,"url":null,"abstract":"<p>Spinocerebellar ataxias (SCAs) are characterized by substantial phenotypic variability. Among them, SCA42 is a rare non-expansion entity presenting with slowly progressive cerebellar syndrome but whose clinical spectrum may be also wider. A 53-year-old male presented with progressive myoclonus-ataxia and intellectual disability. Genetic screening revealed a novel c.3835G > A (p. Asp1279Asn) variant in the <i>CACNA1G</i> gene. SCA42 is a rare non-expansion SCA caused by mutations in <i>CACNA1G</i> on chromosome 17q21, encoding the Ca(V)3.1, a low-threshold voltage-gated T-type calcium channel. The novel variant we identified is potentially involved in channel activity. This case expands the knowledge regarding <i>CACNA1G</i>-associated phenotype and highlights the importance of genetic screening in myoclonus-ataxia disorders.</p>","PeriodicalId":22415,"journal":{"name":"The Cerebellum","volume":"20 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Cerebellum","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s12311-024-01734-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
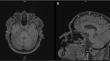
Spinocerebellar ataxias (SCAs) are characterized by substantial phenotypic variability. Among them, SCA42 is a rare non-expansion entity presenting with slowly progressive cerebellar syndrome but whose clinical spectrum may be also wider. A 53-year-old male presented with progressive myoclonus-ataxia and intellectual disability. Genetic screening revealed a novel c.3835G > A (p. Asp1279Asn) variant in the CACNA1G gene. SCA42 is a rare non-expansion SCA caused by mutations in CACNA1G on chromosome 17q21, encoding the Ca(V)3.1, a low-threshold voltage-gated T-type calcium channel. The novel variant we identified is potentially involved in channel activity. This case expands the knowledge regarding CACNA1G-associated phenotype and highlights the importance of genetic screening in myoclonus-ataxia disorders.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
