{"title":"Investigation of Chrysene heterodimers complexes potential energy surface using ab initio computational methods","authors":"Ali Hamzah Alessa","doi":"10.1016/j.comptc.2024.114864","DOIUrl":null,"url":null,"abstract":"<div><p>Numerous chemical and biological entities are greatly impacted by non-covalent interactions in terms of their stability and structure. One such example is the interaction of aromatic rings. These interactions are highly valued in the domains of astrochemistry, biology, chemistry, biochemistry, and material science. The main goals of this study are to explore the Potential Energy Surface (PES) of Chrysene (Chy) heterodimers, identify the most stable configurations among the Chy-Bz, Chy-Np, and Chy-Anth heterodimer complexes, and analyze the inter-molecular interactions between these molecules. This analysis was conducted utilizing <em>ab initio</em> computational techniques. On their PES, the Chy-Np heterodimer exhibited four minima, while the Chy-Bz and Chy-Anth heterodimer complexes showed three. Compared to conformers oriented perpendicularly, co-facial arrangement conformers in Chy heterodimer complexes have stable structures. The global minimum structure of Chy-Bz has been determined to be the face isomer, while Chy-Np and Chy-Anth have global minimum structures of the cross isomer. The binding energies of the structures generated by MP2 are higher than those of DFT-D3, DFT-D4, and SCS-MP2. Optimized geometries and binding energies of larger hydrocarbon aromatic systems are explained in detail by B3LYP-D3 and the recently created B3LYP-D4.</p></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1241 ","pages":"Article 114864"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004031","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/18 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
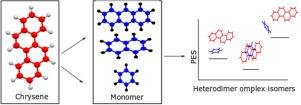
Numerous chemical and biological entities are greatly impacted by non-covalent interactions in terms of their stability and structure. One such example is the interaction of aromatic rings. These interactions are highly valued in the domains of astrochemistry, biology, chemistry, biochemistry, and material science. The main goals of this study are to explore the Potential Energy Surface (PES) of Chrysene (Chy) heterodimers, identify the most stable configurations among the Chy-Bz, Chy-Np, and Chy-Anth heterodimer complexes, and analyze the inter-molecular interactions between these molecules. This analysis was conducted utilizing ab initio computational techniques. On their PES, the Chy-Np heterodimer exhibited four minima, while the Chy-Bz and Chy-Anth heterodimer complexes showed three. Compared to conformers oriented perpendicularly, co-facial arrangement conformers in Chy heterodimer complexes have stable structures. The global minimum structure of Chy-Bz has been determined to be the face isomer, while Chy-Np and Chy-Anth have global minimum structures of the cross isomer. The binding energies of the structures generated by MP2 are higher than those of DFT-D3, DFT-D4, and SCS-MP2. Optimized geometries and binding energies of larger hydrocarbon aromatic systems are explained in detail by B3LYP-D3 and the recently created B3LYP-D4.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
