{"title":"Geometrical features and chemical adsorptions of (Ag3Sn)n clusters","authors":"Qiman Liu , Manli Zhang","doi":"10.1016/j.comptc.2024.114986","DOIUrl":null,"url":null,"abstract":"<div><div>The Ag-Sn alloys are famous ancient intermetallics, with the Ag<sub>3</sub>Sn being a crucial component of the phase diagram. Recently, Ag<sub>3</sub>Sn nanoparticles showcase efficient catalytic CO oxidation capabilities. Here, structural features and stability of (Ag<sub>3</sub>Sn)<sub>n</sub> (n = 1–6) clusters are first analyzed in detail. The results reveal that structures of them evolve from cages to close-packed icosahedra, where Ag are distributed on cores and gradually aggregated, whereas Sn occupy edge positions and become dispersed. Moreover, the icosahedral (Ag<sub>3</sub>Sn)<sub>3</sub> has a higher stability than that of its neighbors and can maintain the structural integrity at 700 K. The molecular orbitals reveal that the (Ag<sub>3</sub>Sn)<sub>3</sub> has an electronic open-shell configuration of 1S<sup>2</sup>1P<sup>6</sup>1D<sup>10</sup>2S<sup>2</sup>1F<sup>1</sup>, which is confirmed by the density of states. Electrostatic potential surfaces show that (Ag<sub>3</sub>Sn)<sub>n</sub> have significant electron-deficient σ-hole regions at Ag sites, which can make C<img>O stretching frequencies and bond lengths have red-shifts. Adsorption energies between (Ag<sub>3</sub>Sn)<sub>n</sub> and CO display odd–even oscillations, ranging from (0.43–0.68) eV, and the direction of charge flows is from CO → clusters. Our work provides inferences to structure evolutions and adsorptions of the Ag<sub>3</sub>Sn alloy at the atomic level.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114986"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24005255","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/12 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
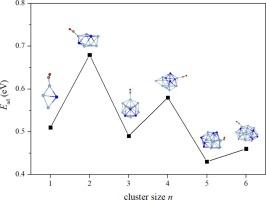
The Ag-Sn alloys are famous ancient intermetallics, with the Ag3Sn being a crucial component of the phase diagram. Recently, Ag3Sn nanoparticles showcase efficient catalytic CO oxidation capabilities. Here, structural features and stability of (Ag3Sn)n (n = 1–6) clusters are first analyzed in detail. The results reveal that structures of them evolve from cages to close-packed icosahedra, where Ag are distributed on cores and gradually aggregated, whereas Sn occupy edge positions and become dispersed. Moreover, the icosahedral (Ag3Sn)3 has a higher stability than that of its neighbors and can maintain the structural integrity at 700 K. The molecular orbitals reveal that the (Ag3Sn)3 has an electronic open-shell configuration of 1S21P61D102S21F1, which is confirmed by the density of states. Electrostatic potential surfaces show that (Ag3Sn)n have significant electron-deficient σ-hole regions at Ag sites, which can make CO stretching frequencies and bond lengths have red-shifts. Adsorption energies between (Ag3Sn)n and CO display odd–even oscillations, ranging from (0.43–0.68) eV, and the direction of charge flows is from CO → clusters. Our work provides inferences to structure evolutions and adsorptions of the Ag3Sn alloy at the atomic level.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
