Development of Tunable Mechanism-Based Carbasugar Ligands that Stabilize Glycoside Hydrolases through the Formation of Transient Covalent Intermediates
Sandeep Bhosale, Sachin Kandalkar, Pierre-André Gilormini, Oluwafemi Akintola, Rhianna Rowland, Pal John Pal Adabala, Dustin King, Matthew C. Deen, Xi Chen, Gideon J. Davies, David J. Vocadlo, Andrew J. Bennet
{"title":"Development of Tunable Mechanism-Based Carbasugar Ligands that Stabilize Glycoside Hydrolases through the Formation of Transient Covalent Intermediates","authors":"Sandeep Bhosale, Sachin Kandalkar, Pierre-André Gilormini, Oluwafemi Akintola, Rhianna Rowland, Pal John Pal Adabala, Dustin King, Matthew C. Deen, Xi Chen, Gideon J. Davies, David J. Vocadlo, Andrew J. Bennet","doi":"10.1021/acscatal.4c04549","DOIUrl":null,"url":null,"abstract":"Mutations in many members of the set of human lysosomal glycoside hydrolases cause a wide range of lysosomal storage diseases. As a result, much effort has been directed toward identifying pharmacological chaperones of these lysosomal enzymes. The majority of the candidate chaperones are active site-directed competitive iminosugar inhibitors but these have met with limited success. As a first step toward an alternative class of pharmacological chaperones we explored the potential of small molecule mechanism-based reversible covalent inhibitors to form transient enzyme–inhibitor adducts. By serial synthesis and kinetic analysis of candidate molecules, we show that rational tuning of the chemical reactivity of glucose-configured carbasugars delivers cyclohexenyl-based allylic carbasugar that react with the lysosomal enzyme β-glucocerebrosidase (GCase) to form covalent enzyme-adducts with different half-lives. X-ray structural analysis of these compounds bound noncovalently to GCase, along with the structures of the covalent adducts of compounds that reacted with the catalytic nucleophile of GCase, reveal unexpected reactivities of these compounds. Using differential scanning fluorimetry, we show that formation of a transient covalent intermediate stabilizes the folded enzyme against thermal denaturation. In addition, these covalent adducts break down to liberate the active enzyme and a product that is no longer inhibitory. We further show that the one compound, which reacts through an unprecedented S<sub>N</sub>1′-like mechanism, exhibits exceptional reactivity–illustrated by this compound also covalently labeling an α-glucosidase. We anticipate that such carbasugar-based single turnover covalent ligands may serve as pharmacological chaperones for lysosomal glycoside hydrolases and other disease-associated retaining glycosidases. The unusual reactivity of these molecules should also open the door to creation of new chemical biology probes to explore the biology of this important superfamily of glycoside hydrolases.","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"30 1","pages":""},"PeriodicalIF":13.1000,"publicationDate":"2024-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acscatal.4c04549","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
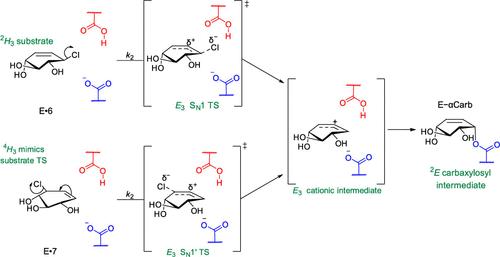
Mutations in many members of the set of human lysosomal glycoside hydrolases cause a wide range of lysosomal storage diseases. As a result, much effort has been directed toward identifying pharmacological chaperones of these lysosomal enzymes. The majority of the candidate chaperones are active site-directed competitive iminosugar inhibitors but these have met with limited success. As a first step toward an alternative class of pharmacological chaperones we explored the potential of small molecule mechanism-based reversible covalent inhibitors to form transient enzyme–inhibitor adducts. By serial synthesis and kinetic analysis of candidate molecules, we show that rational tuning of the chemical reactivity of glucose-configured carbasugars delivers cyclohexenyl-based allylic carbasugar that react with the lysosomal enzyme β-glucocerebrosidase (GCase) to form covalent enzyme-adducts with different half-lives. X-ray structural analysis of these compounds bound noncovalently to GCase, along with the structures of the covalent adducts of compounds that reacted with the catalytic nucleophile of GCase, reveal unexpected reactivities of these compounds. Using differential scanning fluorimetry, we show that formation of a transient covalent intermediate stabilizes the folded enzyme against thermal denaturation. In addition, these covalent adducts break down to liberate the active enzyme and a product that is no longer inhibitory. We further show that the one compound, which reacts through an unprecedented SN1′-like mechanism, exhibits exceptional reactivity–illustrated by this compound also covalently labeling an α-glucosidase. We anticipate that such carbasugar-based single turnover covalent ligands may serve as pharmacological chaperones for lysosomal glycoside hydrolases and other disease-associated retaining glycosidases. The unusual reactivity of these molecules should also open the door to creation of new chemical biology probes to explore the biology of this important superfamily of glycoside hydrolases.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
