Anna V. Pomogaeva, Anna S. Lisovenko, Alexey Y. Timoshkin
{"title":"Facile heterolytic bond splitting of molecular chlorine upon reactions with Lewis bases: Comparison with ICl and I2","authors":"Anna V. Pomogaeva, Anna S. Lisovenko, Alexey Y. Timoshkin","doi":"10.1002/jcc.27507","DOIUrl":null,"url":null,"abstract":"<p>Formation of molecular complexes and subsequent heterolytic halogen-halogen bond splitting upon reactions of molecular Cl<sub>2</sub> with nitrogen-containing Lewis bases (LB) are computationally studied at M06-2X/def2-TZVPD and for selected compounds at CCSD(T)/aug-cc-pvtz//CCSD/aug-cc-pvtz levels of theory. Obtained results are compared with data for ICl and I<sub>2</sub> molecules. Reaction pathways indicate, that in case of Cl<sub>2</sub>∙LB complexes the activation energies for the heterolytic Cl-Cl bond splitting are lower than the activation energies of the homolytic splitting of Cl<sub>2</sub> molecule into chlorine radicals. The heterolytic halogen splitting of molecular complexes of X<sub>2</sub>∙Py with formation of [XPy<sub>2</sub>]<sup>+</sup>…<span></span><math>\n <mrow>\n <msubsup>\n <mi>X</mi>\n <mn>3</mn>\n <mo>−</mo>\n </msubsup>\n </mrow></math> contact ion pairs in the gas phase is slightly endothermic in case of Cl<sub>2</sub> and I<sub>2</sub>, but slightly exothermic in the case of ICl. Formation of {[ClPy<sub>2</sub>]<sup>+</sup>…<span></span><math>\n <mrow>\n <msubsup>\n <mi>Cl</mi>\n <mn>3</mn>\n <mo>−</mo>\n </msubsup>\n </mrow></math>}<sub>2</sub> dimers makes the overall process exothermic. Taking into account that polar solvents favor ionic species, generation of donor-stabilized Cl<sup>+</sup> in the presence of the Lewis bases is expected to be favorable. Thus, in polar solvents the oxidation pathway via donor-stabilized Cl<sup>+</sup> species is viable alternative to the homolytic Cl-Cl bond breaking.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27507","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
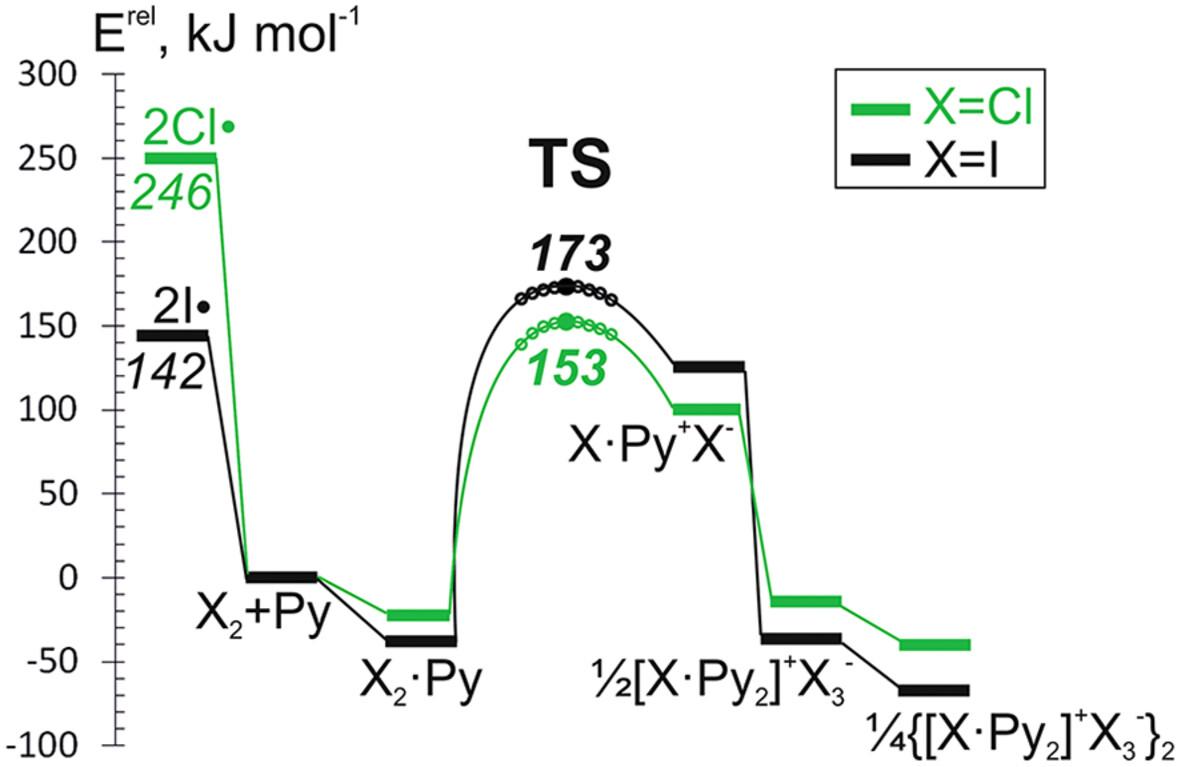
Formation of molecular complexes and subsequent heterolytic halogen-halogen bond splitting upon reactions of molecular Cl2 with nitrogen-containing Lewis bases (LB) are computationally studied at M06-2X/def2-TZVPD and for selected compounds at CCSD(T)/aug-cc-pvtz//CCSD/aug-cc-pvtz levels of theory. Obtained results are compared with data for ICl and I2 molecules. Reaction pathways indicate, that in case of Cl2∙LB complexes the activation energies for the heterolytic Cl-Cl bond splitting are lower than the activation energies of the homolytic splitting of Cl2 molecule into chlorine radicals. The heterolytic halogen splitting of molecular complexes of X2∙Py with formation of [XPy2]+… contact ion pairs in the gas phase is slightly endothermic in case of Cl2 and I2, but slightly exothermic in the case of ICl. Formation of {[ClPy2]+…}2 dimers makes the overall process exothermic. Taking into account that polar solvents favor ionic species, generation of donor-stabilized Cl+ in the presence of the Lewis bases is expected to be favorable. Thus, in polar solvents the oxidation pathway via donor-stabilized Cl+ species is viable alternative to the homolytic Cl-Cl bond breaking.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
