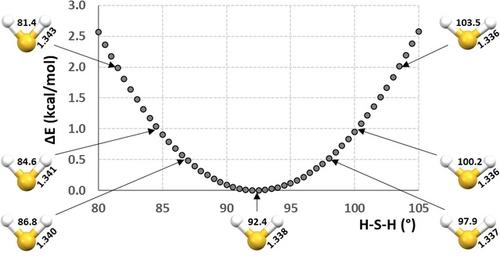
{"title":"How Flexible Is the Hydrogen Sulfide Molecule Structure? Influence of Hydrogen Sulfide Molecule Geometry on Its Hydrogen Bonds.","authors":"Milan R. Milovavnović, Snežana D. Zarić","doi":"10.1002/cplu.202400511","DOIUrl":null,"url":null,"abstract":"<p>The geometry of hydrogen sulfide was studied by calculating potential energy surface (PES) with over 1800 configurations. The calculations were performed at very accurate CCSD(T)/aug-cc-pvz5 level. The most stable geometry on the PES has bond angle (H−S−H) of 92.40° and bond length (S−H) of 1.338 Å. The PES shows that hydrogen sulfide is a quite flexible molecule. Namely, it can change the bonding angle (H−S−H) in the range of 15.6° (from 84.6° to 100.2°) and the bond lengths (S−H) in the range of 0.082 Å (from 1.299 Å to 1.381 Å) with an energy increase of only 1.0 kcal/mol. An influence of hydrogen sulfide geometry on its hydrogen bonds was studied on several hydrogen sulfide/hydrogen sulfide and water/hydrogen sulfide dimers. It showed that the change of hydrogen sulfide geometry does not influence the strength of hydrogen bond. Fully optimized geometries in gas and water solution phases revealed structural differences of both monomers and dimers in gas phase and water phase. SAPT analysis of the optimized dimer geometries showed that in all the dimers electrostatic is the most dominant contribution, while, in the dimers with hydrogen sulfide, the influence of dispersion contribution becomes quite pronounced.</p>","PeriodicalId":148,"journal":{"name":"ChemPlusChem","volume":"90 1","pages":""},"PeriodicalIF":2.8000,"publicationDate":"2024-09-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ChemPlusChem","FirstCategoryId":"92","ListUrlMain":"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cplu.202400511","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
The geometry of hydrogen sulfide was studied by calculating potential energy surface (PES) with over 1800 configurations. The calculations were performed at very accurate CCSD(T)/aug-cc-pvz5 level. The most stable geometry on the PES has bond angle (H−S−H) of 92.40° and bond length (S−H) of 1.338 Å. The PES shows that hydrogen sulfide is a quite flexible molecule. Namely, it can change the bonding angle (H−S−H) in the range of 15.6° (from 84.6° to 100.2°) and the bond lengths (S−H) in the range of 0.082 Å (from 1.299 Å to 1.381 Å) with an energy increase of only 1.0 kcal/mol. An influence of hydrogen sulfide geometry on its hydrogen bonds was studied on several hydrogen sulfide/hydrogen sulfide and water/hydrogen sulfide dimers. It showed that the change of hydrogen sulfide geometry does not influence the strength of hydrogen bond. Fully optimized geometries in gas and water solution phases revealed structural differences of both monomers and dimers in gas phase and water phase. SAPT analysis of the optimized dimer geometries showed that in all the dimers electrostatic is the most dominant contribution, while, in the dimers with hydrogen sulfide, the influence of dispersion contribution becomes quite pronounced.
期刊介绍:
ChemPlusChem is a peer-reviewed, general chemistry journal that brings readers the very best in multidisciplinary research centering on chemistry. It is published on behalf of Chemistry Europe, an association of 16 European chemical societies.
Fully comprehensive in its scope, ChemPlusChem publishes articles covering new results from at least two different aspects (subfields) of chemistry or one of chemistry and one of another scientific discipline (one chemistry topic plus another one, hence the title ChemPlusChem). All suitable submissions undergo balanced peer review by experts in the field to ensure the highest quality, originality, relevance, significance, and validity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
