Sara Tama-Shekan, Valeria Moreno, Ludovic Saba, Chakra P Chaulagain
{"title":"How We Treat Hemolytic Anemia Due to Pyruvate Kinase Deficiency.","authors":"Sara Tama-Shekan, Valeria Moreno, Ludovic Saba, Chakra P Chaulagain","doi":"10.3390/hematolrep16030054","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Pyruvate kinase (PK) deficiency is an inherited red blood cell (RBC) enzyme disorder that results in non-immune chronic hemolytic anemia. Characteristic symptoms of PK deficiency include anemia, fatigue, splenomegaly, jaundice, gallstones, thrombosis, and transfusional iron overload. Previously, treatments aimed at symptomatic management with RBC transfusions, phototherapy, folic acid supplementation, splenectomy, and iron chelation therapy when iron overload was documented. Mitapivat, a recently approved medication for treatment of PK-deficiency hemolytic anemia, is an oral allosteric activator of wild-type and mutant RBC PK enzymes. In this paper, we describe three cases of PK-deficiency anemia treated with mitapivat and describe modern management of this rare hemolytic disorder.</p><p><strong>Methods: </strong>A retrospective healthcare database analysis was conducted to extract relevant information. Both quantitative and qualitative methods were integrated to provide a more comprehensive understanding of the cases.</p><p><strong>Results: </strong>Two patients responded well to treatment with mitapivat, noted by an increase in hemoglobin levels, improvements in hemolytic markers, less frequent or no RBC transfusion requirements, and improvements in fatigue. One patient carrying two non-missense mutations of the <i>PKLR</i> gene did not respond to treatment with mitapivat. As variations in patient-specific factors (including genotype) can lead to different clinical manifestations and responses to treatment, we recommend considering both the phenotype (clinical symptoms and signs) and the genotype of the <i>PKLR</i> gene when making therapeutic decisions about starting a patient on mitapivat.</p><p><strong>Conclusions: </strong>While mitapivat addresses the previously unmet needs of most patients with PK deficiency as the first and only disease-modifying medication to receive approval for this condition, not all patients with PK deficiency are amenable to treatment with mitapivat.</p>","PeriodicalId":12829,"journal":{"name":"Hematology Reports","volume":"16 3","pages":"559-567"},"PeriodicalIF":1.2000,"publicationDate":"2024-08-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11417781/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hematology Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/hematolrep16030054","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Pyruvate kinase (PK) deficiency is an inherited red blood cell (RBC) enzyme disorder that results in non-immune chronic hemolytic anemia. Characteristic symptoms of PK deficiency include anemia, fatigue, splenomegaly, jaundice, gallstones, thrombosis, and transfusional iron overload. Previously, treatments aimed at symptomatic management with RBC transfusions, phototherapy, folic acid supplementation, splenectomy, and iron chelation therapy when iron overload was documented. Mitapivat, a recently approved medication for treatment of PK-deficiency hemolytic anemia, is an oral allosteric activator of wild-type and mutant RBC PK enzymes. In this paper, we describe three cases of PK-deficiency anemia treated with mitapivat and describe modern management of this rare hemolytic disorder.
Methods: A retrospective healthcare database analysis was conducted to extract relevant information. Both quantitative and qualitative methods were integrated to provide a more comprehensive understanding of the cases.
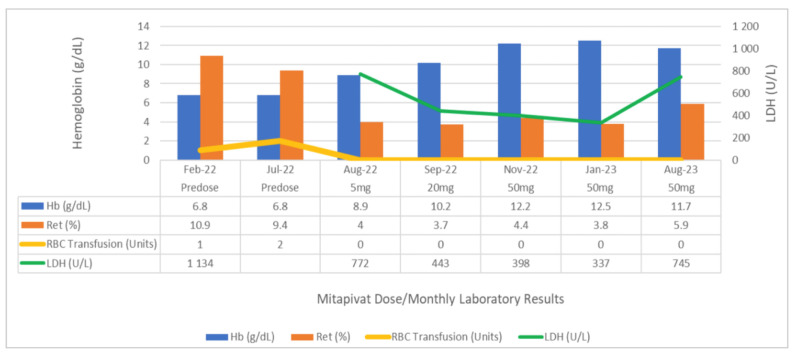
Results: Two patients responded well to treatment with mitapivat, noted by an increase in hemoglobin levels, improvements in hemolytic markers, less frequent or no RBC transfusion requirements, and improvements in fatigue. One patient carrying two non-missense mutations of the PKLR gene did not respond to treatment with mitapivat. As variations in patient-specific factors (including genotype) can lead to different clinical manifestations and responses to treatment, we recommend considering both the phenotype (clinical symptoms and signs) and the genotype of the PKLR gene when making therapeutic decisions about starting a patient on mitapivat.
Conclusions: While mitapivat addresses the previously unmet needs of most patients with PK deficiency as the first and only disease-modifying medication to receive approval for this condition, not all patients with PK deficiency are amenable to treatment with mitapivat.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
