Elena Abati, Delia Gagliardi, Arianna Manini, Roberto Del Bo, Dario Ronchi, Megi Meneri, Francesca Beretta, Annalisa Sarno, Federica Rizzo, Edoardo Monfrini, Alessio Di Fonzo, Maria Teresa Pellecchia, Alberto Brusati, Vincenzo Silani, Giacomo Pietro Comi, Antonia Ratti, Federico Verde, Nicola Ticozzi, Stefania Corti
{"title":"Investigating the prevalence of <i>MFN2</i> mutations in amyotrophic lateral sclerosis: insights from an Italian cohort.","authors":"Elena Abati, Delia Gagliardi, Arianna Manini, Roberto Del Bo, Dario Ronchi, Megi Meneri, Francesca Beretta, Annalisa Sarno, Federica Rizzo, Edoardo Monfrini, Alessio Di Fonzo, Maria Teresa Pellecchia, Alberto Brusati, Vincenzo Silani, Giacomo Pietro Comi, Antonia Ratti, Federico Verde, Nicola Ticozzi, Stefania Corti","doi":"10.1093/braincomms/fcae312","DOIUrl":null,"url":null,"abstract":"<p><p>The <i>MFN2</i> gene encodes mitofusin 2, a key protein for mitochondrial fusion, transport, maintenance and cell communication. <i>MFN2</i> mutations are primarily linked to Charcot-Marie-Tooth disease type 2A. However, a few cases of amyotrophic lateral sclerosis and amyotrophic lateral sclerosis/frontotemporal dementia phenotypes with concomitant <i>MFN2</i> mutations have been previously reported. This study examines the clinical and genetic characteristics of an Italian cohort of amyotrophic lateral sclerosis patients with rare, non-synonymous <i>MFN2</i> mutations. A group of patients (<i>n</i> = 385) diagnosed with amyotrophic lateral sclerosis at our Neurology Units between 2008 and 2023 underwent comprehensive molecular testing, including <i>MFN2</i>. After excluding pathogenic mutations in the main amyotrophic lateral sclerosis-related genes (i.e. <i>C9orf72</i>, <i>SOD1</i>, <i>FUS</i> and <i>TARDBP</i>), <i>MFN2</i> variants were classified based on the American College of Medical Genetics and Genomics guidelines, and demographic and clinical data of <i>MFN2</i>-mutated patients were retrieved. We identified 12 rare, heterozygous, non-synonymous <i>MFN2</i> variants in 19 individuals (4.9%). Eight of these variants, carried by nine patients (2.3%), were either pathogenic, likely pathogenic or variants of unknown significance according to the American College of Medical Genetics and Genomics guidelines. Among these patients, four exhibited a familial pattern of inheritance. The observed phenotypes included classic and bulbar amyotrophic lateral sclerosis, amyotrophic lateral sclerosis/frontotemporal dementia, flail arm, flail leg and progressive muscular atrophy. Median survival after disease onset was extremely variable, ranging from less than 1 to 13 years. This study investigates the prevalence of rare, non-synonymous <i>MFN2</i> variants within an Italian cohort of amyotrophic lateral sclerosis patients, who have been extensively investigated, enhancing our knowledge of the underlying phenotypic spectrum. Further research is needed to understand whether <i>MFN2</i> mutations contribute to motor neuron disease and to what extent. Improving our knowledge regarding the genetic basis of amyotrophic lateral sclerosis is crucial both in a diagnostic and therapeutic perspective.</p>","PeriodicalId":93915,"journal":{"name":"Brain communications","volume":"6 5","pages":"fcae312"},"PeriodicalIF":4.5000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11417610/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain communications","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/braincomms/fcae312","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
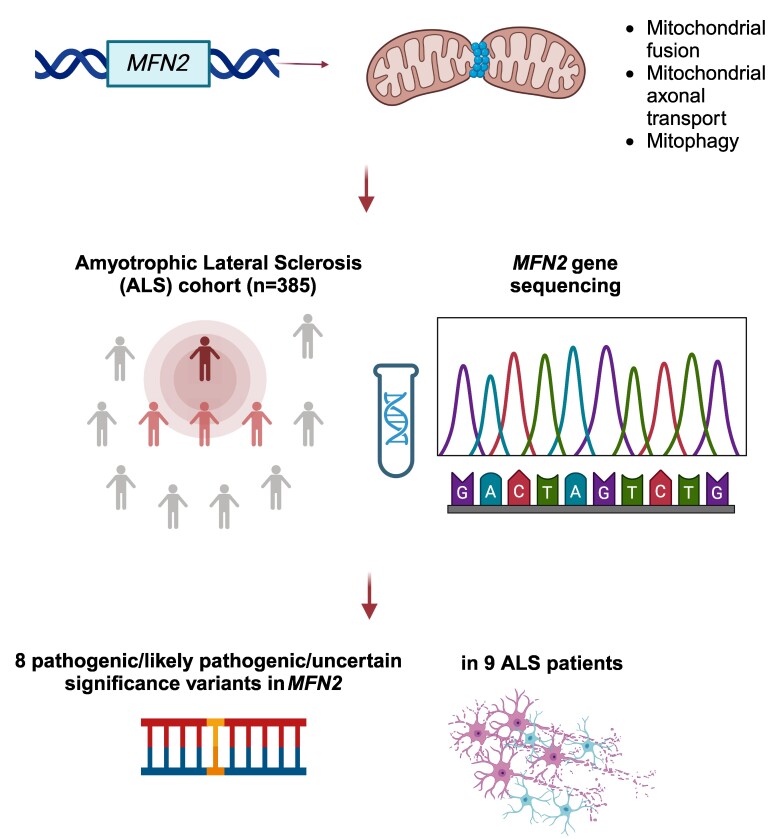
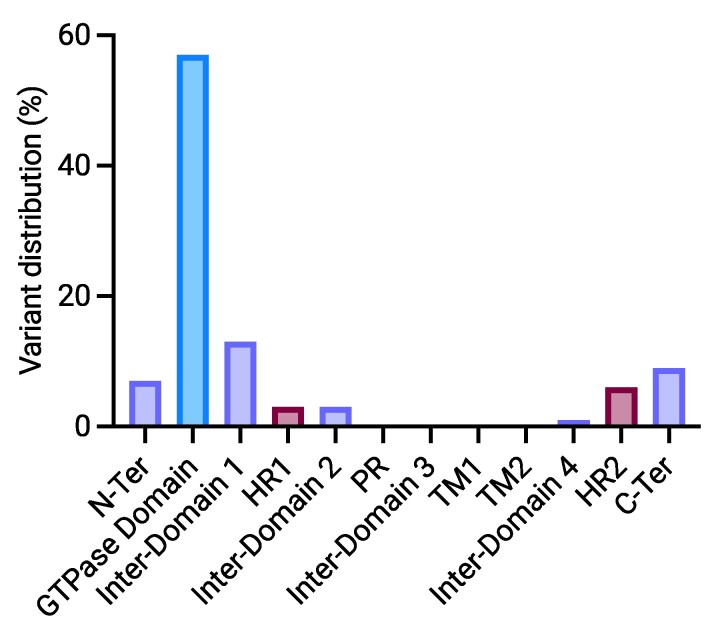
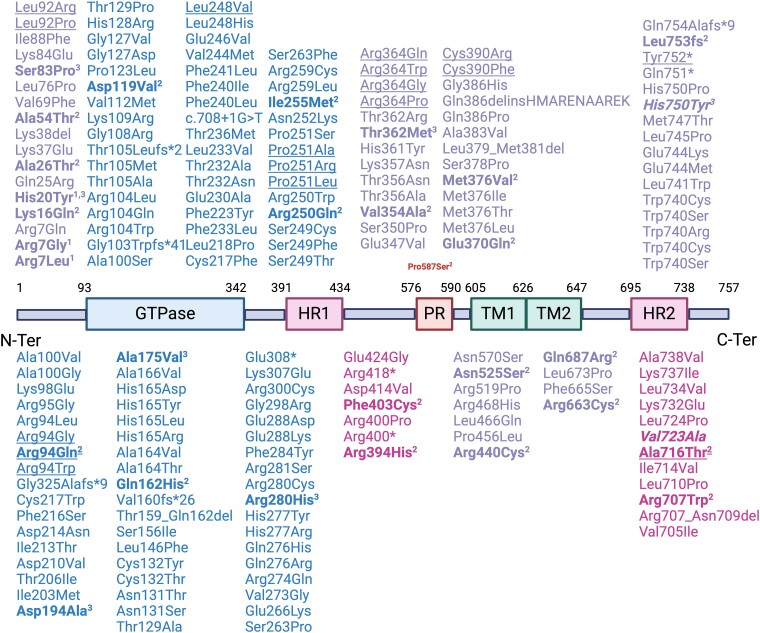
The MFN2 gene encodes mitofusin 2, a key protein for mitochondrial fusion, transport, maintenance and cell communication. MFN2 mutations are primarily linked to Charcot-Marie-Tooth disease type 2A. However, a few cases of amyotrophic lateral sclerosis and amyotrophic lateral sclerosis/frontotemporal dementia phenotypes with concomitant MFN2 mutations have been previously reported. This study examines the clinical and genetic characteristics of an Italian cohort of amyotrophic lateral sclerosis patients with rare, non-synonymous MFN2 mutations. A group of patients (n = 385) diagnosed with amyotrophic lateral sclerosis at our Neurology Units between 2008 and 2023 underwent comprehensive molecular testing, including MFN2. After excluding pathogenic mutations in the main amyotrophic lateral sclerosis-related genes (i.e. C9orf72, SOD1, FUS and TARDBP), MFN2 variants were classified based on the American College of Medical Genetics and Genomics guidelines, and demographic and clinical data of MFN2-mutated patients were retrieved. We identified 12 rare, heterozygous, non-synonymous MFN2 variants in 19 individuals (4.9%). Eight of these variants, carried by nine patients (2.3%), were either pathogenic, likely pathogenic or variants of unknown significance according to the American College of Medical Genetics and Genomics guidelines. Among these patients, four exhibited a familial pattern of inheritance. The observed phenotypes included classic and bulbar amyotrophic lateral sclerosis, amyotrophic lateral sclerosis/frontotemporal dementia, flail arm, flail leg and progressive muscular atrophy. Median survival after disease onset was extremely variable, ranging from less than 1 to 13 years. This study investigates the prevalence of rare, non-synonymous MFN2 variants within an Italian cohort of amyotrophic lateral sclerosis patients, who have been extensively investigated, enhancing our knowledge of the underlying phenotypic spectrum. Further research is needed to understand whether MFN2 mutations contribute to motor neuron disease and to what extent. Improving our knowledge regarding the genetic basis of amyotrophic lateral sclerosis is crucial both in a diagnostic and therapeutic perspective.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
