{"title":"Numerical Investigation of the Quantum Inverse Algorithm on Small Molecules","authors":"Mauro Cainelli*, Reo Baba and Yuki Kurashige*, ","doi":"10.1021/acs.jctc.4c0048310.1021/acs.jctc.4c00483","DOIUrl":null,"url":null,"abstract":"<p >We evaluate the accuracy of the quantum inverse (Q-Inv) algorithm, in which the multiplication of <i>Ĥ</i><sup>–<i>k</i></sup> to the reference wave function is replaced by the Fourier transformed multiplication of e<sup>–iλ<i>Ĥ</i></sup>, as a function of the integration parameters and the iteration power <i>k</i> for various systems, including H<sub>2</sub>, LiH, BeH<sub>2</sub> and the notorious H<sub>4</sub> molecule at square geometry. We further consider the possibility of employing the Gaussian-quadrature rule as an alternate integration method and compared it to the results employing trapezoidal integration. The Q-Inv algorithm is compared to the inverse iteration method using the <i>Ĥ</i><sup>–1</sup> inverse (I-Iter) and the exact inverse by lower-upper decomposition. Energy values are evaluated as the expectation values of the Hamiltonian. Results suggest that the Q-Inv method provides lower energy results than the I-Iter method up to a certain <i>k</i>, after which the energy increases due to errors in the numerical integration that are dependent on the integration interval. A combined Gaussian-quadrature and trapezoidal integration method proved to be more effective at reaching convergence while decreasing the number of operations. For systems like H<sub>4</sub>, in which the Q-Inv cannot reach the expected error threshold, we propose a combination of Q-Inv and I-Iter methods to further decrease the error with <i>k</i> at lower computational cost. Finally, we summarize the recommended procedure when treating unknown systems.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00483","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
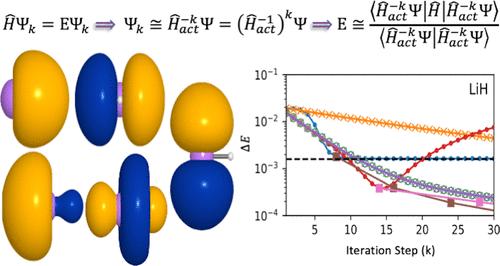
We evaluate the accuracy of the quantum inverse (Q-Inv) algorithm, in which the multiplication of Ĥ–k to the reference wave function is replaced by the Fourier transformed multiplication of e–iλĤ, as a function of the integration parameters and the iteration power k for various systems, including H2, LiH, BeH2 and the notorious H4 molecule at square geometry. We further consider the possibility of employing the Gaussian-quadrature rule as an alternate integration method and compared it to the results employing trapezoidal integration. The Q-Inv algorithm is compared to the inverse iteration method using the Ĥ–1 inverse (I-Iter) and the exact inverse by lower-upper decomposition. Energy values are evaluated as the expectation values of the Hamiltonian. Results suggest that the Q-Inv method provides lower energy results than the I-Iter method up to a certain k, after which the energy increases due to errors in the numerical integration that are dependent on the integration interval. A combined Gaussian-quadrature and trapezoidal integration method proved to be more effective at reaching convergence while decreasing the number of operations. For systems like H4, in which the Q-Inv cannot reach the expected error threshold, we propose a combination of Q-Inv and I-Iter methods to further decrease the error with k at lower computational cost. Finally, we summarize the recommended procedure when treating unknown systems.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
