{"title":"Particle–Particle Random Phase Approximation for Predicting Correlated Excited States of Point Defects","authors":"Jiachen Li*, Yu Jin, Jincheng Yu, Weitao Yang* and Tianyu Zhu*, ","doi":"10.1021/acs.jctc.4c0082910.1021/acs.jctc.4c00829","DOIUrl":null,"url":null,"abstract":"<p >The particle–particle random phase approximation (ppRPA) within the hole–hole channel was recently proposed as an efficient tool for computing excitation energies of point defects in solids [<i>J. Phys. Chem. Lett.</i> 2024, 15, 2757–2764]. In this work, we investigate the application of ppRPA within the particle–particle channel for predicting correlated excited states of point defects, including the carbon-vacancy (VC) in diamond, the oxygen-vacancy (VO) in magnesium oxide (MgO), and the carbon dimer defect (C<sub>B</sub>C<sub>N</sub>) in two-dimensional hexagonal boron nitride (h-BN). Starting from a density functional theory calculation of the (<i>N</i> – 2)-electron ground state, vertical excitation energies of the <i>N</i>-electron system are obtained as the differences between the two-electron addition energies. We show that active-space ppRPA with the B3LYP functional yields accurate excitation energies, with errors mostly smaller than 0.1 eV for tested systems compared to available experimental values. We further develop a natural transition orbital scheme within ppRPA, which provides insights into the multireference character of defect states. This study, together with our previous work, establishes ppRPA as a low-cost and accurate method for investigating excited-state properties of point defect systems.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00829","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
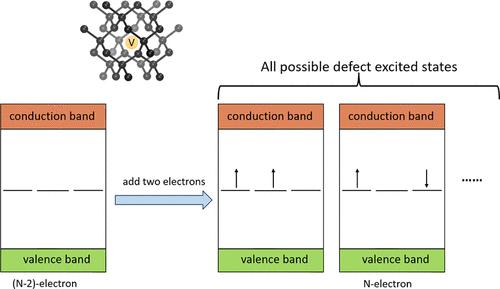
Abstract
The particle–particle random phase approximation (ppRPA) within the hole–hole channel was recently proposed as an efficient tool for computing excitation energies of point defects in solids [J. Phys. Chem. Lett. 2024, 15, 2757–2764]. In this work, we investigate the application of ppRPA within the particle–particle channel for predicting correlated excited states of point defects, including the carbon-vacancy (VC) in diamond, the oxygen-vacancy (VO) in magnesium oxide (MgO), and the carbon dimer defect (CBCN) in two-dimensional hexagonal boron nitride (h-BN). Starting from a density functional theory calculation of the (N – 2)-electron ground state, vertical excitation energies of the N-electron system are obtained as the differences between the two-electron addition energies. We show that active-space ppRPA with the B3LYP functional yields accurate excitation energies, with errors mostly smaller than 0.1 eV for tested systems compared to available experimental values. We further develop a natural transition orbital scheme within ppRPA, which provides insights into the multireference character of defect states. This study, together with our previous work, establishes ppRPA as a low-cost and accurate method for investigating excited-state properties of point defect systems.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
