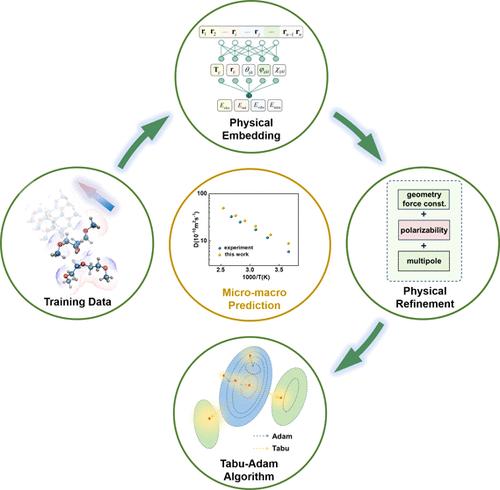
{"title":"Synergistic Integration of Physical Embedding and Machine Learning Enabling Precise and Reliable Force Field","authors":"Lifeng Xu, and , Jian Jiang*, ","doi":"10.1021/acs.jctc.4c0061810.1021/acs.jctc.4c00618","DOIUrl":null,"url":null,"abstract":"<p >Machine-learning force fields have achieved significant strides in accurately reproducing the potential energy surface with quantum chemical accuracy. However, this approach still faces several challenges, e.g., extrapolating to uncharted chemical spaces, interpreting long-range electrostatics, and mapping complex macroscopic properties. To address these issues, we advocate for a synergistic integration of physical principles and machine learning techniques within the framework of a physically informed neural network (PINN). This approach involves incorporating physical knowledge into the parameters of the neural network, coupled with an efficient global optimizer, the Tabu-Adam algorithm, proposed in this work to augment optimization under strict physical constraint. We choose the AMOEBA+ force field as the physics-based model for embedding and then train and test it using the diethylene glycol dimethyl ether (DEGDME) data set as a case study. The results reveal a breakthrough in constructing a precise and noise-robust machine learning force field. Utilizing two training sets with hundreds of samples, our model exhibits remarkable generalization and density functional theory (DFT) accuracy in describing molecular interactions and enables a precise prediction of the macroscopic properties such as the diffusion coefficient with minimal cost. This work provides valuable insight into establishing a fundamental framework of the PINN force field.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00618","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Machine-learning force fields have achieved significant strides in accurately reproducing the potential energy surface with quantum chemical accuracy. However, this approach still faces several challenges, e.g., extrapolating to uncharted chemical spaces, interpreting long-range electrostatics, and mapping complex macroscopic properties. To address these issues, we advocate for a synergistic integration of physical principles and machine learning techniques within the framework of a physically informed neural network (PINN). This approach involves incorporating physical knowledge into the parameters of the neural network, coupled with an efficient global optimizer, the Tabu-Adam algorithm, proposed in this work to augment optimization under strict physical constraint. We choose the AMOEBA+ force field as the physics-based model for embedding and then train and test it using the diethylene glycol dimethyl ether (DEGDME) data set as a case study. The results reveal a breakthrough in constructing a precise and noise-robust machine learning force field. Utilizing two training sets with hundreds of samples, our model exhibits remarkable generalization and density functional theory (DFT) accuracy in describing molecular interactions and enables a precise prediction of the macroscopic properties such as the diffusion coefficient with minimal cost. This work provides valuable insight into establishing a fundamental framework of the PINN force field.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
