{"title":"Multi-Scale Computational Design of Metal–Organic Frameworks for Carbon Capture Using Machine Learning and Multi-Objective Optimization","authors":"Zijun Deng, Lev Sarkisov","doi":"10.1021/acs.chemmater.4c01969","DOIUrl":null,"url":null,"abstract":"In this article, we computationally design a series of metal–organic frameworks (MOFs) optimized for postcombustion carbon capture. Our workflow includes assembling building blocks and topologies into an initial set of hypothetical MOFs, using genetic algorithms to optimize this initial set for high CO<sub>2</sub>/N<sub>2</sub> selectivity, and further evaluating the top materials through process-level modeling of their performance in a modified Skarstrom cycle. We identify two groups of MOFs that exhibit excellent process performance: one with relatively small pores in the range of 3–5 Å and another with larger pores of 6–30 Å. The performance of the first group is driven effectively by the exclusion of N<sub>2</sub> from adsorption, with binding sites able to accommodate only CO<sub>2</sub> molecules. The second group, with larger pores, features binding sites where CO<sub>2</sub> molecules form multiple interactions with oxygen and functional groups of several building blocks, leading to a high CO<sub>2</sub>/N<sub>2</sub> selectivity. Within the employed process model and its assumptions, the materials generated in this study substantially outperform 13X reference zeolites, in silico optimized ion-exchanged LTA zeolites, and CALF-20. While this study does not address the synthesizability, stability, or water interactions of the proposed materials, it marks a significant step forward in developing practical MOFs for carbon capture in three key areas. First, it introduces a generative workflow based on the process-level performance of new materials. Second, it identifies structural features of optimal MOFs for carbon capture, which can serve as design guidelines for future development. Finally, the potential existence of numerous promising materials offers hope that some may progress to laboratory testing and eventual scale-up.","PeriodicalId":33,"journal":{"name":"Chemistry of Materials","volume":null,"pages":null},"PeriodicalIF":7.2000,"publicationDate":"2024-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemistry of Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1021/acs.chemmater.4c01969","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
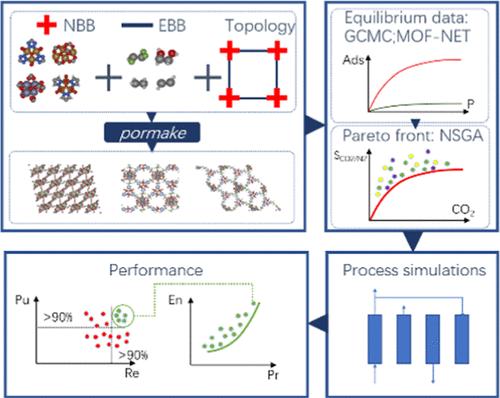
Abstract
In this article, we computationally design a series of metal–organic frameworks (MOFs) optimized for postcombustion carbon capture. Our workflow includes assembling building blocks and topologies into an initial set of hypothetical MOFs, using genetic algorithms to optimize this initial set for high CO2/N2 selectivity, and further evaluating the top materials through process-level modeling of their performance in a modified Skarstrom cycle. We identify two groups of MOFs that exhibit excellent process performance: one with relatively small pores in the range of 3–5 Å and another with larger pores of 6–30 Å. The performance of the first group is driven effectively by the exclusion of N2 from adsorption, with binding sites able to accommodate only CO2 molecules. The second group, with larger pores, features binding sites where CO2 molecules form multiple interactions with oxygen and functional groups of several building blocks, leading to a high CO2/N2 selectivity. Within the employed process model and its assumptions, the materials generated in this study substantially outperform 13X reference zeolites, in silico optimized ion-exchanged LTA zeolites, and CALF-20. While this study does not address the synthesizability, stability, or water interactions of the proposed materials, it marks a significant step forward in developing practical MOFs for carbon capture in three key areas. First, it introduces a generative workflow based on the process-level performance of new materials. Second, it identifies structural features of optimal MOFs for carbon capture, which can serve as design guidelines for future development. Finally, the potential existence of numerous promising materials offers hope that some may progress to laboratory testing and eventual scale-up.
期刊介绍:
The journal Chemistry of Materials focuses on publishing original research at the intersection of materials science and chemistry. The studies published in the journal involve chemistry as a prominent component and explore topics such as the design, synthesis, characterization, processing, understanding, and application of functional or potentially functional materials. The journal covers various areas of interest, including inorganic and organic solid-state chemistry, nanomaterials, biomaterials, thin films and polymers, and composite/hybrid materials. The journal particularly seeks papers that highlight the creation or development of innovative materials with novel optical, electrical, magnetic, catalytic, or mechanical properties. It is essential that manuscripts on these topics have a primary focus on the chemistry of materials and represent a significant advancement compared to prior research. Before external reviews are sought, submitted manuscripts undergo a review process by a minimum of two editors to ensure their appropriateness for the journal and the presence of sufficient evidence of a significant advance that will be of broad interest to the materials chemistry community.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
