Manuel Yáñez, Otilia Mó, M. Merced Montero-Campillo, Ibon Alkorta, José Elguero
{"title":"Hydride and halide abstraction reactions behind the enhanced basicity of Be and Mg clusters with nitrogen bases","authors":"Manuel Yáñez, Otilia Mó, M. Merced Montero-Campillo, Ibon Alkorta, José Elguero","doi":"10.1002/jcc.27509","DOIUrl":null,"url":null,"abstract":"<p>In this study, we investigate the protonation effects on the structure, relative stability and basicity of complexes formed by the interaction of monomers and dimers of BeX<sub>2</sub> and MgX<sub>2</sub> (X = H, F) with NH<sub>3</sub>, CH<sub>2</sub>NH, HCN, and NC<sub>5</sub>H<sub>5</sub> bases. Calculations were performed using the M06-2X/aug-cc-pVTZ formalism, along with QTAIM, ELF and NCI methods for electron density analysis and MBIE and LMO-EDA energy decomposition analyses for interaction enthalpies. The protonation of the MH<sub>2</sub>– and M<sub>2</sub>H<sub>4</sub>–Base complexes occurs at the negatively charged hydrogen atoms of the MH<sub>2</sub> and M<sub>2</sub>H<sub>4</sub> moieties through typical hydride abstraction reactions, while protonation at the N atom of the base is systematically less exothermic. The preference for the hydride transfer mechanism is directly associated with the significant exothermicity of H<sub>2</sub> formation through the interaction between H<sup>−</sup> and H<sup>+</sup>, and the high hydride donor ability of these complexes. The basicity of both, MH<sub>2</sub> and M<sub>2</sub>H<sub>4</sub> compounds increases enormously upon association with the corresponding bases, with the increase exceeding 40 orders of magnitude in terms of ionization constants. Due to the smaller exothermicity of HF formation, the basicity of fluorides is lower than that of hydrides. In Be complexes, the protonation at the N atom of the base dominates over the fluoride abstraction mechanism. However, for the Mg complexes the fluoride abstraction mechanism is energetically the most favorable process, reflecting the greater facility of Mg complexes to lose F<sup>−</sup>.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27509","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27509","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
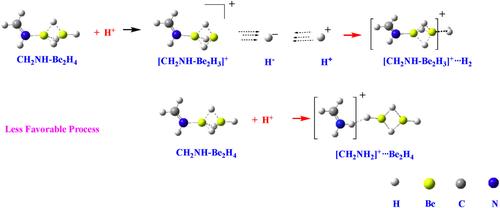
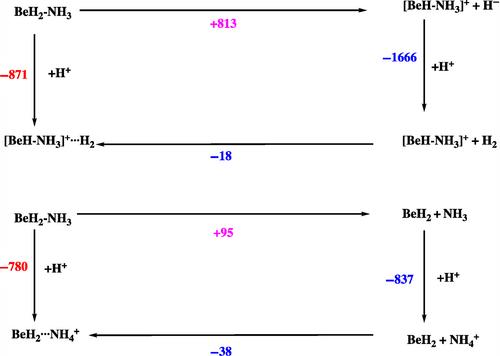
In this study, we investigate the protonation effects on the structure, relative stability and basicity of complexes formed by the interaction of monomers and dimers of BeX2 and MgX2 (X = H, F) with NH3, CH2NH, HCN, and NC5H5 bases. Calculations were performed using the M06-2X/aug-cc-pVTZ formalism, along with QTAIM, ELF and NCI methods for electron density analysis and MBIE and LMO-EDA energy decomposition analyses for interaction enthalpies. The protonation of the MH2– and M2H4–Base complexes occurs at the negatively charged hydrogen atoms of the MH2 and M2H4 moieties through typical hydride abstraction reactions, while protonation at the N atom of the base is systematically less exothermic. The preference for the hydride transfer mechanism is directly associated with the significant exothermicity of H2 formation through the interaction between H− and H+, and the high hydride donor ability of these complexes. The basicity of both, MH2 and M2H4 compounds increases enormously upon association with the corresponding bases, with the increase exceeding 40 orders of magnitude in terms of ionization constants. Due to the smaller exothermicity of HF formation, the basicity of fluorides is lower than that of hydrides. In Be complexes, the protonation at the N atom of the base dominates over the fluoride abstraction mechanism. However, for the Mg complexes the fluoride abstraction mechanism is energetically the most favorable process, reflecting the greater facility of Mg complexes to lose F−.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
