Investigation of structural, elastic, electronic, and optical properties of lead-free double perovskites Cs2XBeBr6 (X = Ge, Sn): a first-principles DFT study
{"title":"Investigation of structural, elastic, electronic, and optical properties of lead-free double perovskites Cs2XBeBr6 (X = Ge, Sn): a first-principles DFT study","authors":"Messaoud Caid, Habib Rached, Djamel Rached, Youcef Rached","doi":"10.1007/s00894-024-06158-x","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>In this study, the structural, elastic, electronic, and optical properties of Cs<sub>2</sub>GeBeBr<sub>6</sub> and Cs<sub>2</sub>SnBeBr<sub>6</sub> halide double perovskites (HDPs) were investigated using density functional theory (DFT) calculations. Notably, the Tran-Blaha modified Becke-Johnson (TB-mBJ) method was employed to predict indirect band gaps of 2.434 eV for Cs<sub>2</sub>GeBeBr<sub>6</sub> and 2.855 eV for Cs<sub>2</sub>SnBeBr<sub>6</sub>. The stability of these compounds in a cubic (Fm-3m) structure was confirmed through formation energy, cohesive energy, tolerance factor, and elastic constants. Furthermore, the ductile nature of the materials was demonstrated through Poisson's and Pugh's ratios. Our optical property analysis, spanning the 0–13 eV energy range, revealed key insights into the dielectric functions, extinction coefficient, electron energy loss, refractive index, optical conductivity, reflectivity, and absorption coefficient. These results highlight the potential of Cs<sub>2</sub>GeBeBr<sub>6</sub> and Cs<sub>2</sub>SnBeBr<sub>6</sub> for future optoelectronic and photovoltaic applications.</p><h3>Methods</h3><p>In this investigation, we employed density functional theory (DFT), implemented using the Wien2k package. The exchange and correlation potentials were treated using the generalized gradient approximation (GGA) and the Tran-Blaha modified Becke-Johnson (TB-mBJ) method. To evaluate dynamic stability, we analyzed the phonon band structures using the CASTEP code.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 10","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-09-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06158-x","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
In this study, the structural, elastic, electronic, and optical properties of Cs2GeBeBr6 and Cs2SnBeBr6 halide double perovskites (HDPs) were investigated using density functional theory (DFT) calculations. Notably, the Tran-Blaha modified Becke-Johnson (TB-mBJ) method was employed to predict indirect band gaps of 2.434 eV for Cs2GeBeBr6 and 2.855 eV for Cs2SnBeBr6. The stability of these compounds in a cubic (Fm-3m) structure was confirmed through formation energy, cohesive energy, tolerance factor, and elastic constants. Furthermore, the ductile nature of the materials was demonstrated through Poisson's and Pugh's ratios. Our optical property analysis, spanning the 0–13 eV energy range, revealed key insights into the dielectric functions, extinction coefficient, electron energy loss, refractive index, optical conductivity, reflectivity, and absorption coefficient. These results highlight the potential of Cs2GeBeBr6 and Cs2SnBeBr6 for future optoelectronic and photovoltaic applications.
Methods
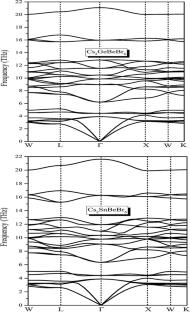
In this investigation, we employed density functional theory (DFT), implemented using the Wien2k package. The exchange and correlation potentials were treated using the generalized gradient approximation (GGA) and the Tran-Blaha modified Becke-Johnson (TB-mBJ) method. To evaluate dynamic stability, we analyzed the phonon band structures using the CASTEP code.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
