Naim Abdul-Khalek, Reinhard Wimmer, Michael Toft Overgaard, Simon Gregersen Echers
{"title":"Decoding the impact of neighboring amino acids on ESI-MS intensity output through deep learning","authors":"Naim Abdul-Khalek, Reinhard Wimmer, Michael Toft Overgaard, Simon Gregersen Echers","doi":"10.1016/j.jprot.2024.105322","DOIUrl":null,"url":null,"abstract":"<div><div>Peptide-level quantification using mass spectrometry (MS) is no trivial task as the physicochemical properties affect both response and detectability. The specific amino acid (AA) sequence affects these properties, however the connection between sequence and intensity output remains poorly understood. In this work, we explore combinations of amino acid pairs (i.e., dimer motifs) to determine a potential relationship between the local amino acid environment and MS1 intensity. For this purpose, a deep learning (DL) model, consisting of an encoder-decoder with an attention mechanism, was built. The attention mechanism allowed to identify the most relevant motifs. Specific patterns were consistently observed where a bulky/aromatic and hydrophobic AA followed by a cationic AA as well as consecutive bulky/aromatic and hydrophobic AAs were found important for the prediction of the MS1 intensity. Correlating attention weights to mean MS1 intensities revealed that some important motifs, particularly containing Trp, His, and Cys, were linked with low responding peptides whereas motifs containing Lys and most bulky hydrophobic AAs were often associated with high responding peptides. Moreover, Asn-Gly was associated with low response. The model predicts MS1 response with a mean average percentage error of ∼11 % and a Pearson correlation coefficient of ∼0.64. While dimer representation of peptide sequences did not improve predictive capacity compared to single AA representation in earlier work, this work adds valuable insight for a better understanding of peptide response in MS analysis.</div></div><div><h3>Significance</h3><div>Mass spectrometry is not inherently quantitative, and the response of a compound relies not only on its concentration but also on the molecular composition. For mass spectrometry-based analysis of peptides, such as in bottom-up proteomics, this directly implies that the response cannot be used directly to quantify individual peptides. Moreover, the dependency of the response on the amino acid sequence of individual peptides remains poorly understood. Using a deep learning model based on a recurrent neural network with an attention mechanism, we here investigate how the presence of dimer motifs within a peptide affects the MS1 response through the analysis of intended equimolar peptide pools comprising almost 200,000 unique peptides in total. Not only do we identify certain dimer classes and specific dimers that substantially affect the MS1 response, but the model is also able to predict peptide intensity with low error rates within the independent test subset. The findings not only improve our understanding of the link between sequence and response for peptides but also highlight the potential of utilizing deep learning for developing methods allowing for absolute, label-free peptide quantification.</div></div>","PeriodicalId":16891,"journal":{"name":"Journal of proteomics","volume":"309 ","pages":"Article 105322"},"PeriodicalIF":2.8000,"publicationDate":"2024-10-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of proteomics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1874391924002549","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/26 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
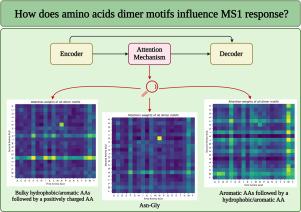
Peptide-level quantification using mass spectrometry (MS) is no trivial task as the physicochemical properties affect both response and detectability. The specific amino acid (AA) sequence affects these properties, however the connection between sequence and intensity output remains poorly understood. In this work, we explore combinations of amino acid pairs (i.e., dimer motifs) to determine a potential relationship between the local amino acid environment and MS1 intensity. For this purpose, a deep learning (DL) model, consisting of an encoder-decoder with an attention mechanism, was built. The attention mechanism allowed to identify the most relevant motifs. Specific patterns were consistently observed where a bulky/aromatic and hydrophobic AA followed by a cationic AA as well as consecutive bulky/aromatic and hydrophobic AAs were found important for the prediction of the MS1 intensity. Correlating attention weights to mean MS1 intensities revealed that some important motifs, particularly containing Trp, His, and Cys, were linked with low responding peptides whereas motifs containing Lys and most bulky hydrophobic AAs were often associated with high responding peptides. Moreover, Asn-Gly was associated with low response. The model predicts MS1 response with a mean average percentage error of ∼11 % and a Pearson correlation coefficient of ∼0.64. While dimer representation of peptide sequences did not improve predictive capacity compared to single AA representation in earlier work, this work adds valuable insight for a better understanding of peptide response in MS analysis.
Significance
Mass spectrometry is not inherently quantitative, and the response of a compound relies not only on its concentration but also on the molecular composition. For mass spectrometry-based analysis of peptides, such as in bottom-up proteomics, this directly implies that the response cannot be used directly to quantify individual peptides. Moreover, the dependency of the response on the amino acid sequence of individual peptides remains poorly understood. Using a deep learning model based on a recurrent neural network with an attention mechanism, we here investigate how the presence of dimer motifs within a peptide affects the MS1 response through the analysis of intended equimolar peptide pools comprising almost 200,000 unique peptides in total. Not only do we identify certain dimer classes and specific dimers that substantially affect the MS1 response, but the model is also able to predict peptide intensity with low error rates within the independent test subset. The findings not only improve our understanding of the link between sequence and response for peptides but also highlight the potential of utilizing deep learning for developing methods allowing for absolute, label-free peptide quantification.
期刊介绍:
Journal of Proteomics is aimed at protein scientists and analytical chemists in the field of proteomics, biomarker discovery, protein analytics, plant proteomics, microbial and animal proteomics, human studies, tissue imaging by mass spectrometry, non-conventional and non-model organism proteomics, and protein bioinformatics. The journal welcomes papers in new and upcoming areas such as metabolomics, genomics, systems biology, toxicogenomics, pharmacoproteomics.
Journal of Proteomics unifies both fundamental scientists and clinicians, and includes translational research. Suggestions for reviews, webinars and thematic issues are welcome.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
