{"title":"Rapid increase of C/EBPα p42 induces growth arrest of acute myeloid leukemia (AML) cells by Cop1 deletion in Trib1-expressing AML","authors":"Yoshitaka Sunami, Seiko Yoshino, Yukari Yamazaki, Takashi Iwamoto, Takuro Nakamura","doi":"10.1038/s41375-024-02430-4","DOIUrl":null,"url":null,"abstract":"Cop1 encodes a ubiquitin E3 ligase that has been well preserved during evolution in both plants and metazoans. In metazoans, the C/EBP family transcription factors are targets for degradation by Cop1, and this process is regulated by the Tribbles pseudokinase family. Over-expression of Tribbles homolog 1 (Trib1) induces acute myeloid leukemia (AML) via Cop1-dependent degradation of the C/EBPα p42 isoform. Here, we induced rapid growth arrest and granulocytic differentiation of Trib1-expressing AML cells using a Cop1 conditional knockout (KO), which is associated with a transient increase in the C/EBPα p42 isoform. The growth-suppressive effect of Cop1 KO was canceled by silencing of Cebpa and reinforced by exogenous expression of the p42 isoform. Moreover, Cop1 KO improved the survival of recipients transplanted with Trib1-expressing AML cells. We further identified a marked increase in Trib1 protein expression in Cop1 KO, indicating that Trib1 is self-degraded by the Cop1 degradosome. COP1 downregulation also inhibits the proliferation of human AML cells in a TRIB1-dependent manner. Taken together, our results provide new insights into the role of Trib1/Cop1 machinery in the C/EBPα p42-dependent leukemogenic activity, and a novel idea to develop new therapeutics.","PeriodicalId":18109,"journal":{"name":"Leukemia","volume":"38 12","pages":"2585-2597"},"PeriodicalIF":13.4000,"publicationDate":"2024-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Leukemia","FirstCategoryId":"3","ListUrlMain":"https://www.nature.com/articles/s41375-024-02430-4","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
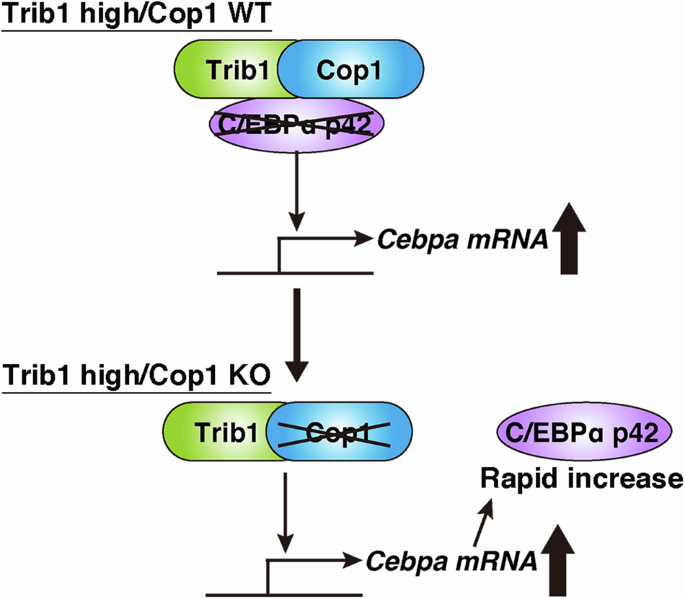
Cop1 encodes a ubiquitin E3 ligase that has been well preserved during evolution in both plants and metazoans. In metazoans, the C/EBP family transcription factors are targets for degradation by Cop1, and this process is regulated by the Tribbles pseudokinase family. Over-expression of Tribbles homolog 1 (Trib1) induces acute myeloid leukemia (AML) via Cop1-dependent degradation of the C/EBPα p42 isoform. Here, we induced rapid growth arrest and granulocytic differentiation of Trib1-expressing AML cells using a Cop1 conditional knockout (KO), which is associated with a transient increase in the C/EBPα p42 isoform. The growth-suppressive effect of Cop1 KO was canceled by silencing of Cebpa and reinforced by exogenous expression of the p42 isoform. Moreover, Cop1 KO improved the survival of recipients transplanted with Trib1-expressing AML cells. We further identified a marked increase in Trib1 protein expression in Cop1 KO, indicating that Trib1 is self-degraded by the Cop1 degradosome. COP1 downregulation also inhibits the proliferation of human AML cells in a TRIB1-dependent manner. Taken together, our results provide new insights into the role of Trib1/Cop1 machinery in the C/EBPα p42-dependent leukemogenic activity, and a novel idea to develop new therapeutics.
期刊介绍:
Title: Leukemia
Journal Overview:
Publishes high-quality, peer-reviewed research
Covers all aspects of research and treatment of leukemia and allied diseases
Includes studies of normal hemopoiesis due to comparative relevance
Topics of Interest:
Oncogenes
Growth factors
Stem cells
Leukemia genomics
Cell cycle
Signal transduction
Molecular targets for therapy
And more
Content Types:
Original research articles
Reviews
Letters
Correspondence
Comments elaborating on significant advances and covering topical issues



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
