{"title":"Bjerrum defects in s-II gas hydrate","authors":"Nevin Uras-Aytemiz , F. Mine Balcı","doi":"10.1016/j.jmgm.2024.108878","DOIUrl":null,"url":null,"abstract":"<div><div>The energy and structure of Bjerrum defects in structure II gas hydrates were investigated by using first-principle calculations for finite-size clusters and periodic 3D lattice systems. The formation energies of these defects were calculated for the first time when the cages of the structure II structure were completely empty and the large cage was filled with a THF molecule. Analogous to findings in ice structures, one of the hydrogen atoms forming the <em>D</em> defect was noted to orient toward the cage. If the excess proton resides in the large cage, it acts as an attraction center for the polar guest molecule, i.e., THF. Therefore, the large cage guest THF molecule stabilizes the <em>D/L</em> defect pair and isolated <em>D/L</em> defect formation energies by forming hydrogen bonds with the <em>D</em> defect. In such cases, the defect structure representing a <em>D/L</em> defect pair containing a THF molecule interacting with one of the hydrogen atoms of the <em>D</em> defect mirrors the guest-induced ones. Notably, the classical Bjerrum defect and the guest-induced Bjerrum defect exhibit a similar phenomenon in defective structures. Contrary to existing literature, it is evident that guest-induced Bjerrum defects involve both the <em>L</em> and <em>D</em> components. The insights gained from this study could potentially offer an alternative perspective to understand various experimental observations, such as those related to dielectric and NMR properties.</div></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"133 ","pages":"Article 108878"},"PeriodicalIF":2.7000,"publicationDate":"2024-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001785","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
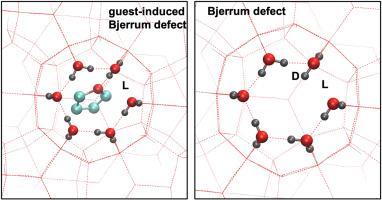
The energy and structure of Bjerrum defects in structure II gas hydrates were investigated by using first-principle calculations for finite-size clusters and periodic 3D lattice systems. The formation energies of these defects were calculated for the first time when the cages of the structure II structure were completely empty and the large cage was filled with a THF molecule. Analogous to findings in ice structures, one of the hydrogen atoms forming the D defect was noted to orient toward the cage. If the excess proton resides in the large cage, it acts as an attraction center for the polar guest molecule, i.e., THF. Therefore, the large cage guest THF molecule stabilizes the D/L defect pair and isolated D/L defect formation energies by forming hydrogen bonds with the D defect. In such cases, the defect structure representing a D/L defect pair containing a THF molecule interacting with one of the hydrogen atoms of the D defect mirrors the guest-induced ones. Notably, the classical Bjerrum defect and the guest-induced Bjerrum defect exhibit a similar phenomenon in defective structures. Contrary to existing literature, it is evident that guest-induced Bjerrum defects involve both the L and D components. The insights gained from this study could potentially offer an alternative perspective to understand various experimental observations, such as those related to dielectric and NMR properties.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
