{"title":"Quantum-accurate machine learning potentials for metal-organic frameworks using temperature driven active learning","authors":"Abhishek Sharma, Stefano Sanvito","doi":"10.1038/s41524-024-01427-y","DOIUrl":null,"url":null,"abstract":"<p>Understanding structural flexibility of metal-organic frameworks (MOFs) via molecular dynamics simulations is crucial to design better MOFs. Density functional theory (DFT) and quantum-chemistry methods provide highly accurate molecular dynamics, but the computational overheads limit their use in long time-dependent simulations. In contrast, classical force fields struggle with the description of coordination bonds. Here we develop a DFT-accurate machine-learning spectral neighbor analysis potentials for two representative MOFs. Their structural and vibrational properties are then studied and tightly compared with available experimental data. Most importantly, we demonstrate an active-learning algorithm, based on mapping the relevant internal coordinates, which drastically reduces the number of training data to be computed at the DFT level. Thus, the workflow presented here appears as an efficient strategy for the study of flexible MOFs with DFT accuracy, but at a fraction of the DFT computational cost.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"12 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01427-y","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
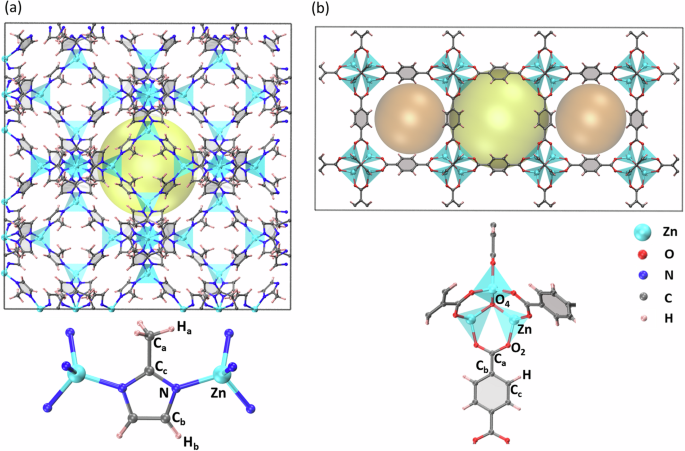
Understanding structural flexibility of metal-organic frameworks (MOFs) via molecular dynamics simulations is crucial to design better MOFs. Density functional theory (DFT) and quantum-chemistry methods provide highly accurate molecular dynamics, but the computational overheads limit their use in long time-dependent simulations. In contrast, classical force fields struggle with the description of coordination bonds. Here we develop a DFT-accurate machine-learning spectral neighbor analysis potentials for two representative MOFs. Their structural and vibrational properties are then studied and tightly compared with available experimental data. Most importantly, we demonstrate an active-learning algorithm, based on mapping the relevant internal coordinates, which drastically reduces the number of training data to be computed at the DFT level. Thus, the workflow presented here appears as an efficient strategy for the study of flexible MOFs with DFT accuracy, but at a fraction of the DFT computational cost.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
