Deanne Nixie R Miao, MacKenzie A P Wilke, John Pham, Feryal Ladha, Mansumeet Singh, Janilyn Arsenio, Emilia Luca, Alain Dabdoub, Wejian Yang, Jun J Yang, Britt I Drögemöller
{"title":"Leveraging large-scale datasets and single cell omics data to develop a polygenic score for cisplatin-induced ototoxicity.","authors":"Deanne Nixie R Miao, MacKenzie A P Wilke, John Pham, Feryal Ladha, Mansumeet Singh, Janilyn Arsenio, Emilia Luca, Alain Dabdoub, Wejian Yang, Jun J Yang, Britt I Drögemöller","doi":"10.1186/s40246-024-00679-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cisplatin-induced ototoxicity (CIO), characterized by irreversible and progressive bilateral hearing loss, is a prevalent adverse effect of cisplatin chemotherapy. Alongside clinical risk factors, genetic variants contribute to CIO and genome-wide association studies (GWAS) have highlighted the polygenicity of this adverse drug reaction. Polygenic scores (PGS), which integrate information from multiple genetic variants across the genome, offer a promising tool for the identification of individuals who are at higher risk for CIO. Integrating large-scale hearing loss GWAS data with single cell omics data holds potential to overcome limitations related to small sample sizes associated with CIO studies, enabling the creation of PGSs to predict CIO risk.</p><p><strong>Results: </strong>We utilized a large-scale hearing loss GWAS and murine inner ear single nuclei RNA-sequencing (snRNA-seq) data to develop two polygenic scores: a hearing loss PGS (PGS<sub>HL</sub>) and a biologically informed PGS for CIO (PGS<sub>CIO</sub>). The PGS<sub>CIO</sub> included only variants which mapped to genes that were differentially expressed within cochlear cells that showed differential abundance in the murine snRNA-seq data post-cisplatin treatment. Evaluation of the association of these PGSs with CIO in our target CIO cohort revealed that PGS<sub>CIO</sub> demonstrated superior performance (P = 5.54 × 10<sup>- 5</sup>) relative to PGS<sub>HL</sub> (P = 2.93 × 10<sup>- 3</sup>). PGS<sub>CIO</sub> was also associated with CIO in our test cohort (P = 0.04), while the PGS<sub>HL</sub> did not show a significant association with CIO (P = 0.52).</p><p><strong>Conclusion: </strong>This study developed the first PGS for CIO using a large-scale hearing loss dataset and a biologically informed filter generated from cisplatin-treated murine inner ear snRNA-seq data. This innovative approach offers new avenues for developing PGSs for pharmacogenomic traits, which could contribute to the implementation of tailored therapeutic interventions. Further, our approach facilitated the identification of specific cochlear cells that may play critical roles in CIO. These novel insights will guide future research aimed at developing targeted therapeutic strategies to prevent CIO.</p>","PeriodicalId":13183,"journal":{"name":"Human Genomics","volume":"18 1","pages":"112"},"PeriodicalIF":4.3000,"publicationDate":"2024-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11463131/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40246-024-00679-5","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Cisplatin-induced ototoxicity (CIO), characterized by irreversible and progressive bilateral hearing loss, is a prevalent adverse effect of cisplatin chemotherapy. Alongside clinical risk factors, genetic variants contribute to CIO and genome-wide association studies (GWAS) have highlighted the polygenicity of this adverse drug reaction. Polygenic scores (PGS), which integrate information from multiple genetic variants across the genome, offer a promising tool for the identification of individuals who are at higher risk for CIO. Integrating large-scale hearing loss GWAS data with single cell omics data holds potential to overcome limitations related to small sample sizes associated with CIO studies, enabling the creation of PGSs to predict CIO risk.
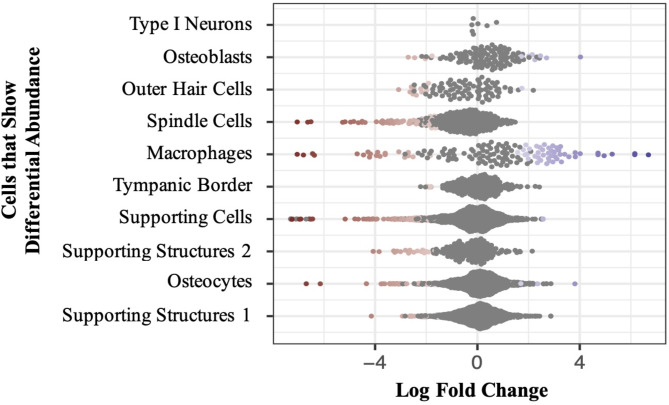
Results: We utilized a large-scale hearing loss GWAS and murine inner ear single nuclei RNA-sequencing (snRNA-seq) data to develop two polygenic scores: a hearing loss PGS (PGSHL) and a biologically informed PGS for CIO (PGSCIO). The PGSCIO included only variants which mapped to genes that were differentially expressed within cochlear cells that showed differential abundance in the murine snRNA-seq data post-cisplatin treatment. Evaluation of the association of these PGSs with CIO in our target CIO cohort revealed that PGSCIO demonstrated superior performance (P = 5.54 × 10- 5) relative to PGSHL (P = 2.93 × 10- 3). PGSCIO was also associated with CIO in our test cohort (P = 0.04), while the PGSHL did not show a significant association with CIO (P = 0.52).
Conclusion: This study developed the first PGS for CIO using a large-scale hearing loss dataset and a biologically informed filter generated from cisplatin-treated murine inner ear snRNA-seq data. This innovative approach offers new avenues for developing PGSs for pharmacogenomic traits, which could contribute to the implementation of tailored therapeutic interventions. Further, our approach facilitated the identification of specific cochlear cells that may play critical roles in CIO. These novel insights will guide future research aimed at developing targeted therapeutic strategies to prevent CIO.
期刊介绍:
Human Genomics is a peer-reviewed, open access, online journal that focuses on the application of genomic analysis in all aspects of human health and disease, as well as genomic analysis of drug efficacy and safety, and comparative genomics.
Topics covered by the journal include, but are not limited to: pharmacogenomics, genome-wide association studies, genome-wide sequencing, exome sequencing, next-generation deep-sequencing, functional genomics, epigenomics, translational genomics, expression profiling, proteomics, bioinformatics, animal models, statistical genetics, genetic epidemiology, human population genetics and comparative genomics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
