Ayşenur Alkaya, Adalet Elçin Yıldız, Esra Bağlan, Semanur Özdel
{"title":"Unveiling the uncommon: diagnostic journey of camurati-engelmann disease in a pediatric patient.","authors":"Ayşenur Alkaya, Adalet Elçin Yıldız, Esra Bağlan, Semanur Özdel","doi":"10.1186/s12969-024-01016-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Camurati-Engelmann disease (CED), also known as progressive diaphyseal dysplasia, is a rare genetic disorder characterized by abnormal thickening of the long bones' diaphysis. This condition is caused by mutations in the transforming growth factor beta-1 (TGFB-1) gene and is typically inherited in an autosomal dominant pattern. Patients with CED often present with symptoms such as chronic bone pain, muscle weakness, fatigue, and difficulty walking.</p><p><strong>Case presentation: </strong>We report a 30-month-old boy who presented with gait abnormality. Initially, toxic synovitis was considered, and non-steroidal anti-inflammatory (NSAİ) treatment was administered. The patient did not respond to NSAİ treatment. Direct radiographs showed diaphyseal thickening, especially in the long bones. Radiologically, CED was suspected, and clinical exome sequencing identified a TGFB-1: c1121C > G (Pro374Arg) heterozygous mutation, which was interpreted as a possible pathogenic variant for CED. A clinical, radiologic, and genetic diagnosis of CED was made.</p><p><strong>Conclusion: </strong>Due to its rarity and variable clinical presentation, the diagnosis of CED can be challenging and often requires a high index of suspicion. Early and accurate diagnosis is crucial for managing symptoms and improving patients' quality of life.</p>","PeriodicalId":54630,"journal":{"name":"Pediatric Rheumatology","volume":"22 1","pages":"89"},"PeriodicalIF":2.3000,"publicationDate":"2024-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11460205/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pediatric Rheumatology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12969-024-01016-9","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Camurati-Engelmann disease (CED), also known as progressive diaphyseal dysplasia, is a rare genetic disorder characterized by abnormal thickening of the long bones' diaphysis. This condition is caused by mutations in the transforming growth factor beta-1 (TGFB-1) gene and is typically inherited in an autosomal dominant pattern. Patients with CED often present with symptoms such as chronic bone pain, muscle weakness, fatigue, and difficulty walking.
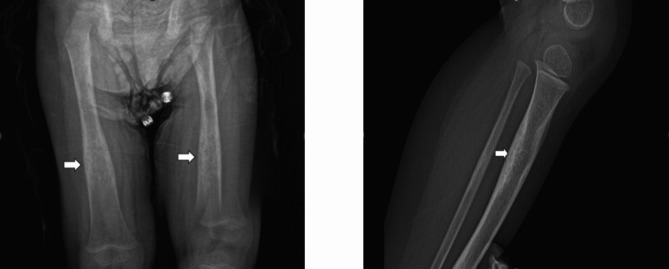
Case presentation: We report a 30-month-old boy who presented with gait abnormality. Initially, toxic synovitis was considered, and non-steroidal anti-inflammatory (NSAİ) treatment was administered. The patient did not respond to NSAİ treatment. Direct radiographs showed diaphyseal thickening, especially in the long bones. Radiologically, CED was suspected, and clinical exome sequencing identified a TGFB-1: c1121C > G (Pro374Arg) heterozygous mutation, which was interpreted as a possible pathogenic variant for CED. A clinical, radiologic, and genetic diagnosis of CED was made.
Conclusion: Due to its rarity and variable clinical presentation, the diagnosis of CED can be challenging and often requires a high index of suspicion. Early and accurate diagnosis is crucial for managing symptoms and improving patients' quality of life.
期刊介绍:
Pediatric Rheumatology is an open access, peer-reviewed, online journal encompassing all aspects of clinical and basic research related to pediatric rheumatology and allied subjects.
The journal’s scope of diseases and syndromes include musculoskeletal pain syndromes, rheumatic fever and post-streptococcal syndromes, juvenile idiopathic arthritis, systemic lupus erythematosus, juvenile dermatomyositis, local and systemic scleroderma, Kawasaki disease, Henoch-Schonlein purpura and other vasculitides, sarcoidosis, inherited musculoskeletal syndromes, autoinflammatory syndromes, and others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
