Marketa Tomkova, Michael John McClellan, Gilles Crevel, Akbar Muhammed Shahid, Nandini Mozumdar, Jakub Tomek, Emelie Shepherd, Sue Cotterill, Benjamin Schuster-Böckler, Skirmantas Kriaucionis
{"title":"Human DNA polymerase ε is a source of C>T mutations at CpG dinucleotides","authors":"Marketa Tomkova, Michael John McClellan, Gilles Crevel, Akbar Muhammed Shahid, Nandini Mozumdar, Jakub Tomek, Emelie Shepherd, Sue Cotterill, Benjamin Schuster-Böckler, Skirmantas Kriaucionis","doi":"10.1038/s41588-024-01945-x","DOIUrl":null,"url":null,"abstract":"C-to-T transitions in CpG dinucleotides are the most prevalent mutations in human cancers and genetic diseases. These mutations have been attributed to deamination of 5-methylcytosine (5mC), an epigenetic modification found on CpGs. We recently linked CpG>TpG mutations to replication and hypothesized that errors introduced by polymerase ε (Pol ε) may represent an alternative source of mutations. Here we present a new method called polymerase error rate sequencing (PER-seq) to measure the error spectrum of DNA polymerases in isolation. We find that the most common human cancer-associated Pol ε mutant (P286R) produces an excess of CpG>TpG errors, phenocopying the mutation spectrum of tumors carrying this mutation and deficiencies in mismatch repair. Notably, we also discover that wild-type Pol ε has a sevenfold higher error rate when replicating 5mCpG compared to C in other contexts. Together, our results from PER-seq and human cancers demonstrate that replication errors are a major contributor to CpG>TpG mutagenesis in replicating cells, fundamentally changing our understanding of this important disease-causing mutational mechanism. A new method called polymerase error rate sequencing (PER-seq) can measure the nucleotide misincorporation rate of DNA polymerases. DNA polymerase ε mutants produce an excess of CpG","PeriodicalId":18985,"journal":{"name":"Nature genetics","volume":"56 11","pages":"2506-2516"},"PeriodicalIF":29.0000,"publicationDate":"2024-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s41588-024-01945-x.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41588-024-01945-x","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
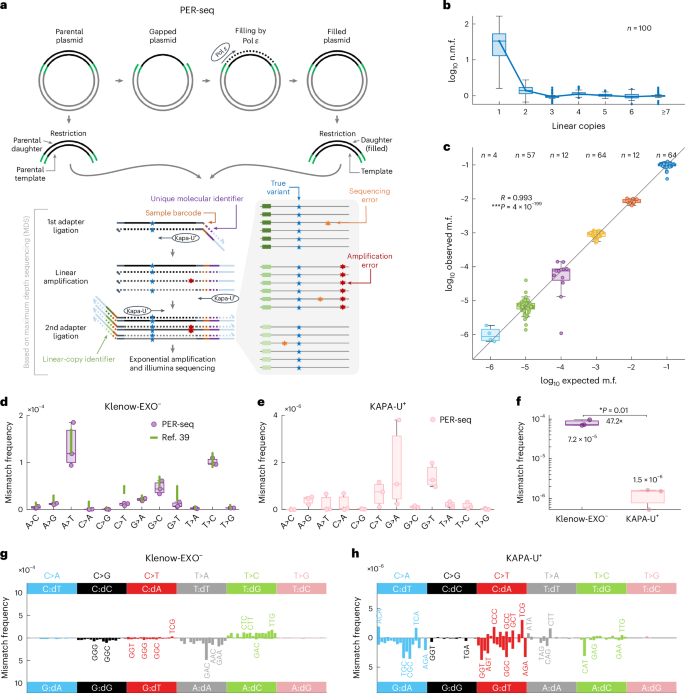
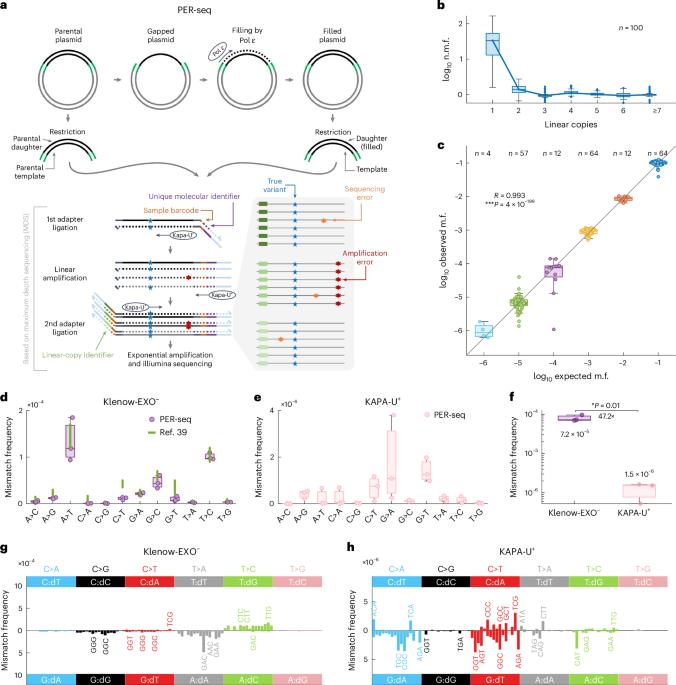
C-to-T transitions in CpG dinucleotides are the most prevalent mutations in human cancers and genetic diseases. These mutations have been attributed to deamination of 5-methylcytosine (5mC), an epigenetic modification found on CpGs. We recently linked CpG>TpG mutations to replication and hypothesized that errors introduced by polymerase ε (Pol ε) may represent an alternative source of mutations. Here we present a new method called polymerase error rate sequencing (PER-seq) to measure the error spectrum of DNA polymerases in isolation. We find that the most common human cancer-associated Pol ε mutant (P286R) produces an excess of CpG>TpG errors, phenocopying the mutation spectrum of tumors carrying this mutation and deficiencies in mismatch repair. Notably, we also discover that wild-type Pol ε has a sevenfold higher error rate when replicating 5mCpG compared to C in other contexts. Together, our results from PER-seq and human cancers demonstrate that replication errors are a major contributor to CpG>TpG mutagenesis in replicating cells, fundamentally changing our understanding of this important disease-causing mutational mechanism. A new method called polymerase error rate sequencing (PER-seq) can measure the nucleotide misincorporation rate of DNA polymerases. DNA polymerase ε mutants produce an excess of CpG
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
