Xifeng Fu, Zhi-Ying Zhao, Sai Guo, Zi-Ang Nan, Lingyi Meng, Can-Zhong Lu
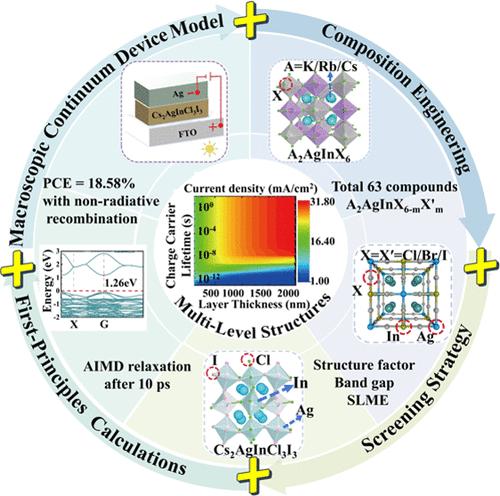
{"title":"Enhancing Optoelectronic Performance of All-Inorganic Double Perovskites via Halogen Doping: Synergistic Screening Strategies and Multiscale Simulations.","authors":"Xifeng Fu, Zhi-Ying Zhao, Sai Guo, Zi-Ang Nan, Lingyi Meng, Can-Zhong Lu","doi":"10.1021/acs.jctc.4c01115","DOIUrl":null,"url":null,"abstract":"<p><p>Designing all-inorganic double perovskites through element mixing is a promising strategy to enhance their optoelectronic performance and structural stability. The complex interplay between multilevel structures and optoelectronic properties in element-mixed double perovskites necessitates further in-depth theoretical exploration. In this study, we employ screening strategies and multiscale simulations combining first-principles methods and device-scale continuum models to identify two novel element-mixed compounds, Rb<sub>2</sub>AgInCl<sub>3</sub>I<sub>3</sub> and Cs<sub>2</sub>AgInCl<sub>3</sub>I<sub>3</sub>, as promising candidates for photovoltaic applications. These compounds exhibit favorable structural factors and suitable direct band gaps. Theoretical investigations using first-principles methods with the HSE06 functional reveal direct band gaps of 0.98 and 1.26 eV for Rb<sub>2</sub>AgInCl<sub>3</sub>I<sub>3</sub> and Cs<sub>2</sub>AgInCl<sub>3</sub>I<sub>3</sub>, respectively, with corresponding optical absorption coefficients exceeding 10<sup>5</sup> cm<sup>-1</sup> in the visible light range. Cs<sub>2</sub>AgInCl<sub>3</sub>I<sub>3</sub> features high charge mobilities of approximately 20 cm<sup>2</sup>·V<sup>-1</sup>·s<sup>-1</sup> and a notable single-junction spectroscopic limited maximum efficiency (SLME) of 25.54%. Further analysis using the device-scale continuum model simulated the nonradiative recombination effects on power conversion efficiency, integrating quantum-mechanically calculated optoelectronic properties. These theoretical investigations, which bridge composition engineering with multiscale simulations, provide valuable insights into screening novel, lead-free, halogen<b>-</b>mixed double metal perovskite optoelectronic devices, highlighting their potential for high-performance solar energy applications.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"9148-9160"},"PeriodicalIF":5.5000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01115","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/11 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Designing all-inorganic double perovskites through element mixing is a promising strategy to enhance their optoelectronic performance and structural stability. The complex interplay between multilevel structures and optoelectronic properties in element-mixed double perovskites necessitates further in-depth theoretical exploration. In this study, we employ screening strategies and multiscale simulations combining first-principles methods and device-scale continuum models to identify two novel element-mixed compounds, Rb2AgInCl3I3 and Cs2AgInCl3I3, as promising candidates for photovoltaic applications. These compounds exhibit favorable structural factors and suitable direct band gaps. Theoretical investigations using first-principles methods with the HSE06 functional reveal direct band gaps of 0.98 and 1.26 eV for Rb2AgInCl3I3 and Cs2AgInCl3I3, respectively, with corresponding optical absorption coefficients exceeding 105 cm-1 in the visible light range. Cs2AgInCl3I3 features high charge mobilities of approximately 20 cm2·V-1·s-1 and a notable single-junction spectroscopic limited maximum efficiency (SLME) of 25.54%. Further analysis using the device-scale continuum model simulated the nonradiative recombination effects on power conversion efficiency, integrating quantum-mechanically calculated optoelectronic properties. These theoretical investigations, which bridge composition engineering with multiscale simulations, provide valuable insights into screening novel, lead-free, halogen-mixed double metal perovskite optoelectronic devices, highlighting their potential for high-performance solar energy applications.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
