Regina M. Burganova , Zafari Umar , Oleg V. Nedopekin , Ilya V. Chepkasov , Irina I. Piyanzina
{"title":"Complex investigation of XF3(X = Gd, Tb, Dy, Ho and Er) fluorides under pressure: An ab-initio perspective","authors":"Regina M. Burganova , Zafari Umar , Oleg V. Nedopekin , Ilya V. Chepkasov , Irina I. Piyanzina","doi":"10.1016/j.commatsci.2024.113428","DOIUrl":null,"url":null,"abstract":"<div><div>Comprehensive systematic density functional theory calculations were performed for five typical rare earth trifluorides, namely GdF<sub>3</sub>, TbF<sub>3</sub>, DyF<sub>3</sub>, HoF<sub>3</sub>, and ErF<sub>3</sub>, under pressures up to 30<!--> <!-->GPa, demonstrating induced phase transitions in agreement with available experimental observations. For the first time the careful check of simulation routine is performed for the selected set of rare earth trifluorides. An extensive selection of the methodology parameters revealed different behaviors for the systems under study. Based on comparative analysis with available experimental data, suitable computation details were suggested for further calculations. For the selected trifluorides, the evolution of lattice parameters and volume, criteria of stability, and elastic stiffness coefficients were analyzed with pressure, which also were calculated for the first time. Additionally, electronic, magnetic, and optical features were captured within the scope of the work for all five compounds in two phases, along with a comparative analysis with experimental data where available.</div></div>","PeriodicalId":10650,"journal":{"name":"Computational Materials Science","volume":"246 ","pages":"Article 113428"},"PeriodicalIF":3.3000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Materials Science","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0927025624006499","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/8 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
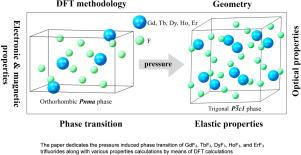
Comprehensive systematic density functional theory calculations were performed for five typical rare earth trifluorides, namely GdF3, TbF3, DyF3, HoF3, and ErF3, under pressures up to 30 GPa, demonstrating induced phase transitions in agreement with available experimental observations. For the first time the careful check of simulation routine is performed for the selected set of rare earth trifluorides. An extensive selection of the methodology parameters revealed different behaviors for the systems under study. Based on comparative analysis with available experimental data, suitable computation details were suggested for further calculations. For the selected trifluorides, the evolution of lattice parameters and volume, criteria of stability, and elastic stiffness coefficients were analyzed with pressure, which also were calculated for the first time. Additionally, electronic, magnetic, and optical features were captured within the scope of the work for all five compounds in two phases, along with a comparative analysis with experimental data where available.
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
