{"title":"Nrf2 deficiency in muscle attenuates experimental autoimmune myositis-induced muscle weakness","authors":"Koichi Himori, Mami Yamada, Takahiro Onoki, Daisuke Matsumaru, Hozumi Motohashi, Mitsuharu Okutsu","doi":"10.1113/JP286534","DOIUrl":null,"url":null,"abstract":"<div>\n \n <section>\n \n \n <div>Idiopathic inflammatory myopathies (IIMs) are systemic autoimmune diseases characterised by muscle weakness. Although multiple physiological and pathological processes are associated with IIMs, T-lymphocyte infiltration into muscle plays a key role in the development and exacerbation of IIMs. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor that regulates inflammatory responses; therefore, muscle Nrf2 may serve an important role in the development of IIMs. In this study, we demonstrated that experimental autoimmune myositis (EAM) causes loss of muscle mass and function in oxidative and glycolytic muscles in C57BL/6 mice. EAM increased CD4<sup>+</sup> and CD8<sup>+</sup> T-lymphocyte infiltration, as well as interferon-gamma (IFN-γ) and tumour necrosis factor-alpha (TNF-α) mRNA expression in oxidative soleus and glycolytic extensor digitorum longus muscles, along with elevated chemokine mRNA levels (i.e. CCL3, CCL5, CXCL9, CXCL10 and CXCL16). IFN-γ and TNF-α treatments increased the mRNA expression levels of these chemokines in C2C12 myotubes. EAM also increased phosphorylated Nrf2 at Ser40 in soleus and glycolytic white vastus lateralis muscle. Although the expression of several chemokines was affected by Nrf2 activation following tert-butylhydroquinone treatment or Keap1 knockdown, CCL5 mRNA expression significantly increased in C2C12 myotubes and mouse skeletal muscle. Moreover, muscle-specific Nrf2 knockout in mice attenuates EAM-induced loss of muscle mass and function, which was associated with the inhibition of CCL5 mRNA expression, CD8<sup>+</sup> T-lymphocyte infiltration and IFN-γ mRNA expression. Collectively, these findings reveal that regulating Nrf2 activity is a promising therapeutic approach for treating IIM-mediated muscle weakness.\n\n <figure>\n <div><picture>\n <source></source></picture><p></p>\n </div>\n </figure>\n </div>\n </section>\n \n <section>\n \n <h3> Key points</h3>\n \n <div>\n <ul>\n \n <li>Experimental autoimmune myositis (EAM) causes loss of muscle mass and function.</li>\n \n <li>Loss of muscle mass and function in EAM were associated with increased chemokine mRNA expression (i.e. CCL3, CCL5, CXCL9, CXCL10 and CXCL16), T-lymphocyte infiltration and inflammatory cytokine mRNA expression (i.e. IFN-γ and TNF-α) in the skeletal muscle.</li>\n \n <li>EAM activated Nrf2 in muscle and increased Nrf2 activity <i>in vivo</i> and <i>in vitro</i> increased CCL5 mRNA expression.</li>\n \n <li>Muscle-specific Nrf2 knockout in mice attenuated EAM-induced muscle weakness by inhibiting CCL5 mRNA expression, CD8<sup>+</sup> T-lymphocyte migration and IFN-γ mRNA expression in muscles.</li>\n \n <li>These results provide further evidence for the potential therapeutic targeting of Nrf2 to mitigate EAM-induced muscle weakness.</li>\n </ul>\n </div>\n </section>\n </div>","PeriodicalId":50088,"journal":{"name":"Journal of Physiology-London","volume":"602 22","pages":"6189-6207"},"PeriodicalIF":4.4000,"publicationDate":"2024-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physiology-London","FirstCategoryId":"3","ListUrlMain":"https://physoc.onlinelibrary.wiley.com/doi/10.1113/JP286534","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
Idiopathic inflammatory myopathies (IIMs) are systemic autoimmune diseases characterised by muscle weakness. Although multiple physiological and pathological processes are associated with IIMs, T-lymphocyte infiltration into muscle plays a key role in the development and exacerbation of IIMs. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor that regulates inflammatory responses; therefore, muscle Nrf2 may serve an important role in the development of IIMs. In this study, we demonstrated that experimental autoimmune myositis (EAM) causes loss of muscle mass and function in oxidative and glycolytic muscles in C57BL/6 mice. EAM increased CD4+ and CD8+ T-lymphocyte infiltration, as well as interferon-gamma (IFN-γ) and tumour necrosis factor-alpha (TNF-α) mRNA expression in oxidative soleus and glycolytic extensor digitorum longus muscles, along with elevated chemokine mRNA levels (i.e. CCL3, CCL5, CXCL9, CXCL10 and CXCL16). IFN-γ and TNF-α treatments increased the mRNA expression levels of these chemokines in C2C12 myotubes. EAM also increased phosphorylated Nrf2 at Ser40 in soleus and glycolytic white vastus lateralis muscle. Although the expression of several chemokines was affected by Nrf2 activation following tert-butylhydroquinone treatment or Keap1 knockdown, CCL5 mRNA expression significantly increased in C2C12 myotubes and mouse skeletal muscle. Moreover, muscle-specific Nrf2 knockout in mice attenuates EAM-induced loss of muscle mass and function, which was associated with the inhibition of CCL5 mRNA expression, CD8+ T-lymphocyte infiltration and IFN-γ mRNA expression. Collectively, these findings reveal that regulating Nrf2 activity is a promising therapeutic approach for treating IIM-mediated muscle weakness.
Key points
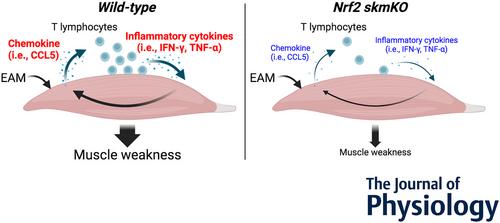
Experimental autoimmune myositis (EAM) causes loss of muscle mass and function.
Loss of muscle mass and function in EAM were associated with increased chemokine mRNA expression (i.e. CCL3, CCL5, CXCL9, CXCL10 and CXCL16), T-lymphocyte infiltration and inflammatory cytokine mRNA expression (i.e. IFN-γ and TNF-α) in the skeletal muscle.
EAM activated Nrf2 in muscle and increased Nrf2 activity in vivo and in vitro increased CCL5 mRNA expression.
Muscle-specific Nrf2 knockout in mice attenuated EAM-induced muscle weakness by inhibiting CCL5 mRNA expression, CD8+ T-lymphocyte migration and IFN-γ mRNA expression in muscles.
These results provide further evidence for the potential therapeutic targeting of Nrf2 to mitigate EAM-induced muscle weakness.
期刊介绍:
The Journal of Physiology publishes full-length original Research Papers and Techniques for Physiology, which are short papers aimed at disseminating new techniques for physiological research. Articles solicited by the Editorial Board include Perspectives, Symposium Reports and Topical Reviews, which highlight areas of special physiological interest. CrossTalk articles are short editorial-style invited articles framing a debate between experts in the field on controversial topics. Letters to the Editor and Journal Club articles are also published. All categories of papers are subjected to peer reivew.
The Journal of Physiology welcomes submitted research papers in all areas of physiology. Authors should present original work that illustrates new physiological principles or mechanisms. Papers on work at the molecular level, at the level of the cell membrane, single cells, tissues or organs and on systems physiology are all acceptable. Theoretical papers and papers that use computational models to further our understanding of physiological processes will be considered if based on experimentally derived data and if the hypothesis advanced is directly amenable to experimental testing. While emphasis is on human and mammalian physiology, work on lower vertebrate or invertebrate preparations may be suitable if it furthers the understanding of the functioning of other organisms including mammals.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
