Yuxin Chang , Ian Benlolo , Yang Bai , Christoff Reimer , Daojin Zhou , Hengrui Zhang , Hidetoshi Matsumura , Hitarth Choubisa , Xiao-Yan Li , Wei Chen , Pengfei Ou , Isaac Tamblyn , Edward H. Sargent
{"title":"High-entropy alloy electrocatalysts screened using machine learning informed by quantum-inspired similarity analysis","authors":"Yuxin Chang , Ian Benlolo , Yang Bai , Christoff Reimer , Daojin Zhou , Hengrui Zhang , Hidetoshi Matsumura , Hitarth Choubisa , Xiao-Yan Li , Wei Chen , Pengfei Ou , Isaac Tamblyn , Edward H. Sargent","doi":"10.1016/j.matt.2024.10.001","DOIUrl":null,"url":null,"abstract":"<div><div>The discovery of new electrocatalysts can be aided by density functional theory (DFT) computation of overpotentials based on the energies of chemical intermediates on prospective adsorption sites. We hypothesize that when training a machine learning model on DFT data, one could improve accuracy by introducing a quantitative measure of similarity among adsorption sites. When we augment a graph neural network-based machine learning workflow using similarity as an input feature, we find that the required training dataset size is decreased from 1,600 to 800, leading to a 2× acceleration: the number of DFT calculations required to train to a given level of accuracy is cut in half. This approach identifies Fe<sub>0.125</sub>Co<sub>0.125</sub>Ni<sub>0.229</sub>Ir<sub>0.229</sub>Ru<sub>0.292</sub> as a promising oxygen reduction reaction catalyst with an overpotential of 0.24 V, outperforming a Pt/C benchmark. We examine, by studying experimentally four additional HEAs, the predictive power of the computational approach.</div></div>","PeriodicalId":388,"journal":{"name":"Matter","volume":"7 11","pages":"Pages 4099-4113"},"PeriodicalIF":17.5000,"publicationDate":"2024-11-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Matter","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2590238524005289","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
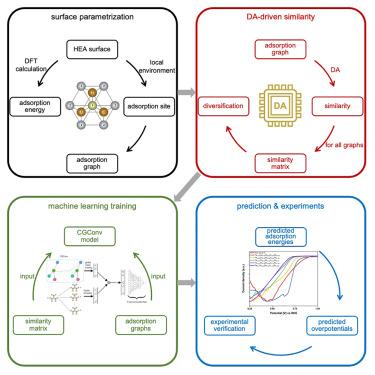
The discovery of new electrocatalysts can be aided by density functional theory (DFT) computation of overpotentials based on the energies of chemical intermediates on prospective adsorption sites. We hypothesize that when training a machine learning model on DFT data, one could improve accuracy by introducing a quantitative measure of similarity among adsorption sites. When we augment a graph neural network-based machine learning workflow using similarity as an input feature, we find that the required training dataset size is decreased from 1,600 to 800, leading to a 2× acceleration: the number of DFT calculations required to train to a given level of accuracy is cut in half. This approach identifies Fe0.125Co0.125Ni0.229Ir0.229Ru0.292 as a promising oxygen reduction reaction catalyst with an overpotential of 0.24 V, outperforming a Pt/C benchmark. We examine, by studying experimentally four additional HEAs, the predictive power of the computational approach.
期刊介绍:
Matter, a monthly journal affiliated with Cell, spans the broad field of materials science from nano to macro levels,covering fundamentals to applications. Embracing groundbreaking technologies,it includes full-length research articles,reviews, perspectives,previews, opinions, personnel stories, and general editorial content.
Matter aims to be the primary resource for researchers in academia and industry, inspiring the next generation of materials scientists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
