{"title":"Crystal Structure Prediction Using Generative Adversarial Network with Data-Driven Latent Space Fusion Strategy","authors":"Zian Chen, Haichao Li, Chen Zhang, Hongbin Zhang, Yongxiao Zhao, Jian Cao, Tao He, Lina Xu, Hongping Xiao, Yi Li, Hezhu Shao, Xiaoyu Yang, Xiao He, Guoyong Fang","doi":"10.1021/acs.jctc.4c01096","DOIUrl":null,"url":null,"abstract":"Crystal structure prediction (CSP) is an important field of material design. Herein, we propose a novel generative adversarial network model, guided by a data-driven approach and incorporating the real physical structure of crystals, to address the complexity of high-dimensional data and improve prediction accuracy in materials science. The model, termed GAN-DDLSF, introduces a novel sampling method called data-driven latent space fusion (DDLSF), which aims to optimize the latent space of generative adversarial networks (GANs) by combining the statistical properties of real data with a standard Gaussian distribution, effectively mitigating the “mode collapse” problem prevalent in GANs. Our approach introduces a more refined generation mechanism specifically for binary crystal structures such as gallium nitride (GaN). By optimizing for the specific crystallographic features of GaN while maintaining structural rationality, we achieve higher precision and efficiency in predicting and designing structures for this particular material system. The model generates 9321 GaN binary crystal structures, with 16.59% reaching a stable state and 24.21% found to be metastable. These results can significantly enhance the accuracy of crystal structure predictions and provide valuable insights into the potential of the GAN-DDLSF approach for the discovery and design of binary, ternary, and multinary materials, offering new perspectives and methods for materials science research and applications.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"59 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01096","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
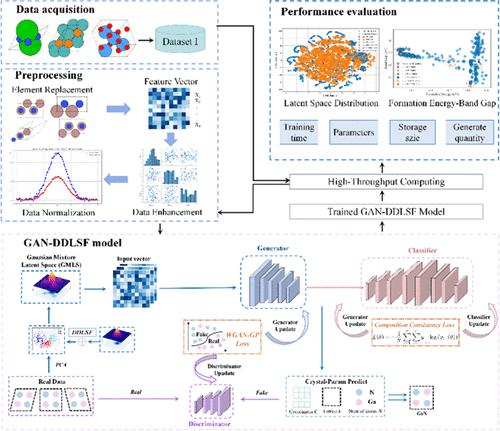
Crystal structure prediction (CSP) is an important field of material design. Herein, we propose a novel generative adversarial network model, guided by a data-driven approach and incorporating the real physical structure of crystals, to address the complexity of high-dimensional data and improve prediction accuracy in materials science. The model, termed GAN-DDLSF, introduces a novel sampling method called data-driven latent space fusion (DDLSF), which aims to optimize the latent space of generative adversarial networks (GANs) by combining the statistical properties of real data with a standard Gaussian distribution, effectively mitigating the “mode collapse” problem prevalent in GANs. Our approach introduces a more refined generation mechanism specifically for binary crystal structures such as gallium nitride (GaN). By optimizing for the specific crystallographic features of GaN while maintaining structural rationality, we achieve higher precision and efficiency in predicting and designing structures for this particular material system. The model generates 9321 GaN binary crystal structures, with 16.59% reaching a stable state and 24.21% found to be metastable. These results can significantly enhance the accuracy of crystal structure predictions and provide valuable insights into the potential of the GAN-DDLSF approach for the discovery and design of binary, ternary, and multinary materials, offering new perspectives and methods for materials science research and applications.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
