W. M. Uvin G. De Alwis, K. L. Dimuthu M. Weerawardene, Kevin L. Shuford
{"title":"Highly Efficient Solar Energy Harvesting in Phosphorene–Graphene Quantum Dot van der Waals Heterostructures: An Ab Initio Approach","authors":"W. M. Uvin G. De Alwis, K. L. Dimuthu M. Weerawardene, Kevin L. Shuford","doi":"10.1021/acs.jpcc.4c04286","DOIUrl":null,"url":null,"abstract":"This study delves into the intricacies of creating highly effective power conversion assemblies from van der Waals heterostructures of phosphorene and graphene quantum dots by employing density functional theory calculations. We emphasize the role of individual monomer properties and their interlayer interactions in the power conversion ability by focusing on visible light absorption, charge carrier generation, and their separation. Different edge atom functionalization (H, NH<sub>2</sub>, Cl, OCN, CN) in the phosphorene quantum dots and heteroatom doping (Group IV = Si, Ge, and Group VI = O, S, Se) in the basal plane of graphene quantum dots were employed to alter the monomer properties. Our results indicate that select combinations of these modification techniques yield staggered frontier molecular orbital alignments (Type II) with spatial separation of the highest occupied and lowest unoccupied molecular orbitals. These candidates possess an improved visible light absorption range with reduced intensities owing to the dominance of charge transfer excitations. Edge functionalization of phosphorene was identified as the most significant contributor to interlayer interaction strength, with functional groups that are electron-withdrawing in nature forming stronger interactions. Heteroatom doping of graphene was recognized as the most important contributor to improving visible light absorbance owing to the reduction in fundamental gaps. From the candidates considered, systems with relatively weaker interlayer interactions were determined to be better at charge carrier separation due to the potential gradient being concentrated at the interfacial region. These systems possess approximated power conversion efficiencies ranging between 11 and 29%, among the highest reported for quantum dot systems characterized by density functional theory calculations.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"26 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-10-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c04286","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
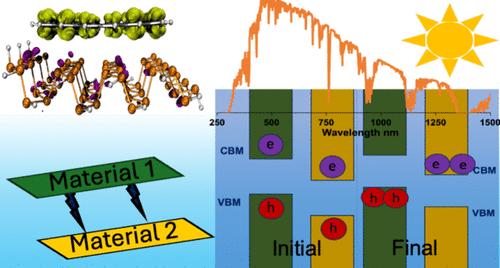
This study delves into the intricacies of creating highly effective power conversion assemblies from van der Waals heterostructures of phosphorene and graphene quantum dots by employing density functional theory calculations. We emphasize the role of individual monomer properties and their interlayer interactions in the power conversion ability by focusing on visible light absorption, charge carrier generation, and their separation. Different edge atom functionalization (H, NH2, Cl, OCN, CN) in the phosphorene quantum dots and heteroatom doping (Group IV = Si, Ge, and Group VI = O, S, Se) in the basal plane of graphene quantum dots were employed to alter the monomer properties. Our results indicate that select combinations of these modification techniques yield staggered frontier molecular orbital alignments (Type II) with spatial separation of the highest occupied and lowest unoccupied molecular orbitals. These candidates possess an improved visible light absorption range with reduced intensities owing to the dominance of charge transfer excitations. Edge functionalization of phosphorene was identified as the most significant contributor to interlayer interaction strength, with functional groups that are electron-withdrawing in nature forming stronger interactions. Heteroatom doping of graphene was recognized as the most important contributor to improving visible light absorbance owing to the reduction in fundamental gaps. From the candidates considered, systems with relatively weaker interlayer interactions were determined to be better at charge carrier separation due to the potential gradient being concentrated at the interfacial region. These systems possess approximated power conversion efficiencies ranging between 11 and 29%, among the highest reported for quantum dot systems characterized by density functional theory calculations.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
