Tomoyasu Higashimoto, Martin E Garber, Lauren Hipp, Jenna Damon, Qing Li
{"title":"Atypical Presentation of Congenital Insensitivity to Pain With Anhidrosis Leading to Diagnostic Odyssey.","authors":"Tomoyasu Higashimoto, Martin E Garber, Lauren Hipp, Jenna Damon, Qing Li","doi":"10.1002/mgg3.70027","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Congenital insensitivity to pain with anhidrosis (CIPA) (OMIM 256800) is a rare autosomal-recessive condition, also known as hereditary sensory and autonomic neuropathy type IV (HSAN-IV). The most commonly reported features include anhidrosis, intellectual disability, self-mutilation, febrile episodes, impaired temperature perception, recurrent infections and/or autonomic nervous system impairment. Major joint destruction and joint deformity known as Charcot (neuropathic) joints are also seen in CIPA patients attributed to insensitivity to joint pain.</p><p><strong>Methods: </strong>We present a case of a 46-year-old female affected with CIPA with a known NTRK1 variant and previously unidentified variant. Minigene reporter constructs were generated encompassing the exon 8 to exon 13 of the NTRK1 gene using the reference sequence and one harboring c.1483 + 5G > A variant identified in our proband. Minigene constructs were transfected into HEK293T cells, and the transcript was analysed for splicing to evaluate the effect of this variant in splicing.</p><p><strong>Results: </strong>The patient (46-year-old female) exhibited right ankle joint deformity around 5 years of age. Patient also experienced lumbar compression and knee damage in adulthood. She had undergone a significant number of evaluations without clear diagnosis. Her presentation lacked many of the common clinical presentations of CIPA, and therefore, the focus of her evaluation was directed towards her unexplained joint deformities. Exome sequencing revealed a known pathogenic variant in NTRK1 (c.851 - 33T > A:p.? [Intron 7]) and a novel NTRK1 variant (c.1483 + 5G > A:p.? [Intron 11]), which was later re-classified as likely pathogenic. The patient was started on a biologic disease-modifying anti-rheumatic medication (bDMARD) due to a possible inflammatory etiology of her joint deformity. Molecular diagnosis allowed for modification of her treatment and surveillance strategies. Our minigene splicing assay demonstrated that the presence of the c.1483 + 5G > A variant has a negative effect on splicing, supporting the pathogenicity of this novel variant.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"12 10","pages":"e70027"},"PeriodicalIF":1.6000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11513606/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70027","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Congenital insensitivity to pain with anhidrosis (CIPA) (OMIM 256800) is a rare autosomal-recessive condition, also known as hereditary sensory and autonomic neuropathy type IV (HSAN-IV). The most commonly reported features include anhidrosis, intellectual disability, self-mutilation, febrile episodes, impaired temperature perception, recurrent infections and/or autonomic nervous system impairment. Major joint destruction and joint deformity known as Charcot (neuropathic) joints are also seen in CIPA patients attributed to insensitivity to joint pain.
Methods: We present a case of a 46-year-old female affected with CIPA with a known NTRK1 variant and previously unidentified variant. Minigene reporter constructs were generated encompassing the exon 8 to exon 13 of the NTRK1 gene using the reference sequence and one harboring c.1483 + 5G > A variant identified in our proband. Minigene constructs were transfected into HEK293T cells, and the transcript was analysed for splicing to evaluate the effect of this variant in splicing.
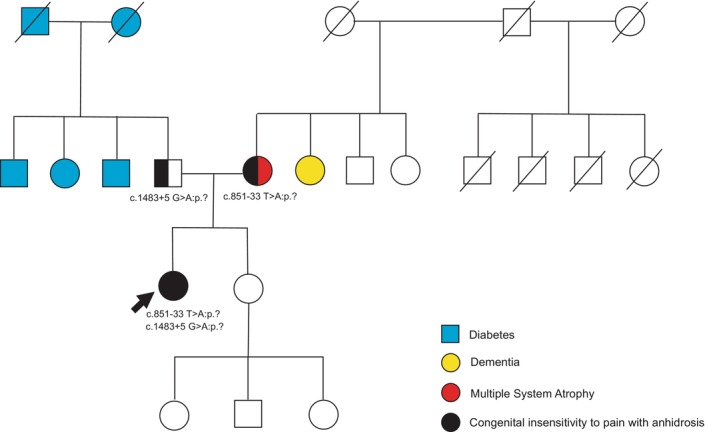
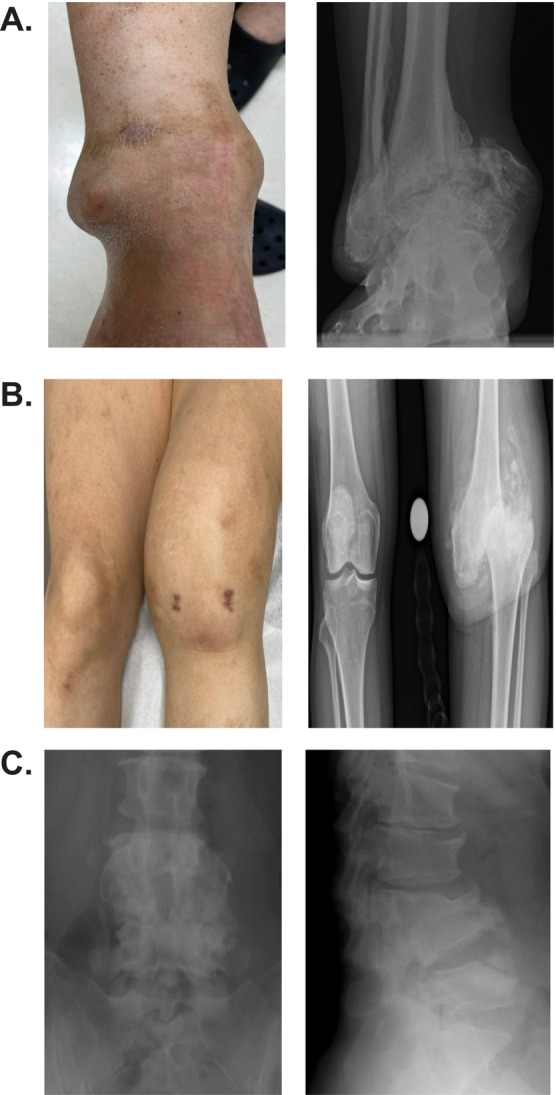
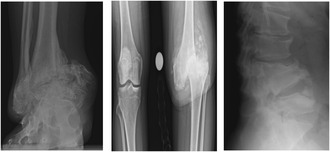
Results: The patient (46-year-old female) exhibited right ankle joint deformity around 5 years of age. Patient also experienced lumbar compression and knee damage in adulthood. She had undergone a significant number of evaluations without clear diagnosis. Her presentation lacked many of the common clinical presentations of CIPA, and therefore, the focus of her evaluation was directed towards her unexplained joint deformities. Exome sequencing revealed a known pathogenic variant in NTRK1 (c.851 - 33T > A:p.? [Intron 7]) and a novel NTRK1 variant (c.1483 + 5G > A:p.? [Intron 11]), which was later re-classified as likely pathogenic. The patient was started on a biologic disease-modifying anti-rheumatic medication (bDMARD) due to a possible inflammatory etiology of her joint deformity. Molecular diagnosis allowed for modification of her treatment and surveillance strategies. Our minigene splicing assay demonstrated that the presence of the c.1483 + 5G > A variant has a negative effect on splicing, supporting the pathogenicity of this novel variant.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
