{"title":"Evaluate the potential adsorption of graphynes (perfect and doped) for nitrogen mustard gas: A first principles study","authors":"Shu Zijing","doi":"10.1016/j.comptc.2024.114911","DOIUrl":null,"url":null,"abstract":"<div><div>Density functional theory (DFT) calculations are used to thoroughly examine the reactivity and electronic sensitivity of pristine and BN-doped graphyne (BNG) toward nitrogen mustard (NM). Graphyne’s electrical conductivity is unaffected by the weak adsorption of NM, which occurs via the Cl atom on the material with an adsorption energy of roughly −3.1 kcal.mol<sup>−1</sup>. In addition to decreasing graphyne’s reactivity and work function, substituting isoelectronic <img>B<img>N<img> linkages for <img>C<img>C<img> linkages enhances the HOMO-LUMO energy gap (Eg). BNG’s electrical conductivity increases when Eg drops from 2.99 to 1.82 eV due to the adsorption of NM. Additionally, a significant change in BNG’s work function results in a variation in the field electron emission current. Lastly, it is anticipated that the desorption of NM from the BNG surface will take a brief recovery time of roughly 0.05 s at room temperature. It has also been demonstrated that NM concentration affects changes in electrical conductivity. The findings also suggest that BNG could be a promising NM sensor.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1241 ","pages":"Article 114911"},"PeriodicalIF":3.0000,"publicationDate":"2024-10-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X2400450X","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
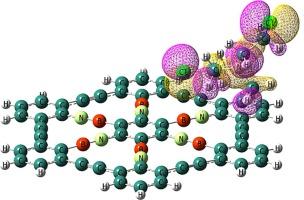
Abstract
Density functional theory (DFT) calculations are used to thoroughly examine the reactivity and electronic sensitivity of pristine and BN-doped graphyne (BNG) toward nitrogen mustard (NM). Graphyne’s electrical conductivity is unaffected by the weak adsorption of NM, which occurs via the Cl atom on the material with an adsorption energy of roughly −3.1 kcal.mol−1. In addition to decreasing graphyne’s reactivity and work function, substituting isoelectronic BN linkages for CC linkages enhances the HOMO-LUMO energy gap (Eg). BNG’s electrical conductivity increases when Eg drops from 2.99 to 1.82 eV due to the adsorption of NM. Additionally, a significant change in BNG’s work function results in a variation in the field electron emission current. Lastly, it is anticipated that the desorption of NM from the BNG surface will take a brief recovery time of roughly 0.05 s at room temperature. It has also been demonstrated that NM concentration affects changes in electrical conductivity. The findings also suggest that BNG could be a promising NM sensor.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
