Yuwei Zhang, Nadire Nayir, Yun Kyung Shin, Qian Mao, Ga-Un Jeong, Chen Chen, Joan M. Redwing, Adri C. T. van Duin
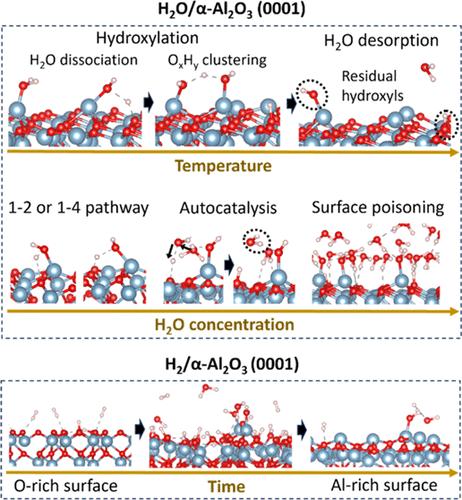
{"title":"ReaxFF Study of Surface Chemical Reactions between α-Al2O3 Substrates and H2O/H2 Gas-Phase Molecules","authors":"Yuwei Zhang, Nadire Nayir, Yun Kyung Shin, Qian Mao, Ga-Un Jeong, Chen Chen, Joan M. Redwing, Adri C. T. van Duin","doi":"10.1021/acs.jpcc.4c04669","DOIUrl":null,"url":null,"abstract":"We developed an Al/O/H ReaxFF force field to explore chemical reactions on α-Al<sub>2</sub>O<sub>3</sub> surfaces in H<sub>2</sub>O/H<sub>2</sub> gas-phase environments. This force field generates surface energy profiles of A-, C-, R-, and M-planes with various terminations (Al- or O-) and predicts the thermodynamic and kinetic behaviors of hydrolysis on Al-terminated α-Al<sub>2</sub>O<sub>3</sub> (0001), consistent with quantum chemical studies. Molecular dynamics (MD) simulations of H<sub>2</sub>O/α-Al<sub>2</sub>O<sub>3</sub> (0001) reveal that water autocatalysis plays a significant role in accelerating H<sub>2</sub>O dissociations on Al-terminated α-Al<sub>2</sub>O<sub>3</sub> (0001). Compared with the 50% Al-terminated surface, the 100% Al-terminated surface becomes more easily hydroxylated at temperatures as low as 350 K, relying more on an O<i><sub>x</sub></i>H<i><sub>y</sub></i> clustering mechanism than complete H<sub>2</sub>O dissociations, and desorbs significantly more H<sub>2</sub>O molecules once heated up to 500 K or higher. But heating cannot eliminate surface hydroxyls for either case, and achieving a Gibbsite-like surface by H<sub>2</sub>O exposure is unlikely. H<sub>2</sub>O dissociations on α-Al<sub>2</sub>O<sub>3</sub> (0001) terminated with randomly distributed surface Al species deviate from 1–2 and 1–4 pathways due to irregular vacancy defects, and a random surface appears to be more reactive to H<sub>2</sub>O than the ordered one with the same surface Al coverage. Simulations of H<sub>2</sub>/α-Al<sub>2</sub>O<sub>3</sub> suggest that the combination of a dense surface O coverage and a low thermodynamic surface stability leads to elevated H<sub>2</sub> dissociation kinetics. To accelerate the surface O removals of 100% O-terminated α-Al<sub>2</sub>O<sub>3</sub> (0001) in H<sub>2</sub> gas exposure, we reduced the H–H σ bond energy parameter, equivalent to lowering the H<sub>2</sub> dissociation barrier by ∼ 19.4 kcal/mol during the simulation. After ∼ 1.5 ns, the surface termination became comparable to the 100% Al-terminated one but retained a small quantity of hydroxyls. This force field reveals how the α-Al<sub>2</sub>O<sub>3</sub> crystallographic plane and the surface termination influence the dissociation behaviors of H<sub>2</sub>O/H<sub>2</sub> gas molecules and lays the foundation for future force field developments targeted at thin film epitaxy on sapphire.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"11 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-10-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c04669","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
We developed an Al/O/H ReaxFF force field to explore chemical reactions on α-Al2O3 surfaces in H2O/H2 gas-phase environments. This force field generates surface energy profiles of A-, C-, R-, and M-planes with various terminations (Al- or O-) and predicts the thermodynamic and kinetic behaviors of hydrolysis on Al-terminated α-Al2O3 (0001), consistent with quantum chemical studies. Molecular dynamics (MD) simulations of H2O/α-Al2O3 (0001) reveal that water autocatalysis plays a significant role in accelerating H2O dissociations on Al-terminated α-Al2O3 (0001). Compared with the 50% Al-terminated surface, the 100% Al-terminated surface becomes more easily hydroxylated at temperatures as low as 350 K, relying more on an OxHy clustering mechanism than complete H2O dissociations, and desorbs significantly more H2O molecules once heated up to 500 K or higher. But heating cannot eliminate surface hydroxyls for either case, and achieving a Gibbsite-like surface by H2O exposure is unlikely. H2O dissociations on α-Al2O3 (0001) terminated with randomly distributed surface Al species deviate from 1–2 and 1–4 pathways due to irregular vacancy defects, and a random surface appears to be more reactive to H2O than the ordered one with the same surface Al coverage. Simulations of H2/α-Al2O3 suggest that the combination of a dense surface O coverage and a low thermodynamic surface stability leads to elevated H2 dissociation kinetics. To accelerate the surface O removals of 100% O-terminated α-Al2O3 (0001) in H2 gas exposure, we reduced the H–H σ bond energy parameter, equivalent to lowering the H2 dissociation barrier by ∼ 19.4 kcal/mol during the simulation. After ∼ 1.5 ns, the surface termination became comparable to the 100% Al-terminated one but retained a small quantity of hydroxyls. This force field reveals how the α-Al2O3 crystallographic plane and the surface termination influence the dissociation behaviors of H2O/H2 gas molecules and lays the foundation for future force field developments targeted at thin film epitaxy on sapphire.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
