{"title":"Exploring Free Energy Landscapes for Protein Partitioning into Membrane Domains in All-Atom and Coarse-Grained Simulations.","authors":"Seulki Kwon, Ayan Majumder, John E Straub","doi":"10.1021/acs.jctc.4c00881","DOIUrl":null,"url":null,"abstract":"<p><p>It is known that membrane environment can impact the structure and function of integral membrane proteins. As such, elucidation of the thermodynamic driving forces governing protein partitioning between membrane domains of varying lipid composition is a fundamental topic in membrane biophysics. Molecular dynamics simulations provide valuable tools for quantitatively characterizing the free energy landscapes governing protein partitioning at the molecular level. In this study, we propose an efficient simulation methodology for the calculation of free energies for the partitioning of transmembrane proteins between liquid-disorder (<i>L</i><sub>d</sub>) and liquid-ordered (<i>L</i><sub>o</sub>) domains in all-atom (AA) phase-separated lipid bilayers. The computed potential of mean force defining the equilibrium partition coefficients is compared for AA and coarse-grained systems. Energy decomposition is used to identify differences in the underlying thermodynamics. Our findings highlight the importance of employing AA models to accurately estimate relevant free energy changes during protein translation between membrane domains.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"9687-9698"},"PeriodicalIF":5.5000,"publicationDate":"2024-11-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00881","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/1 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
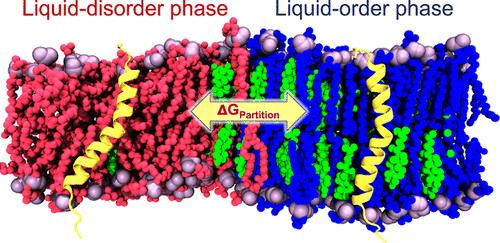
Abstract
It is known that membrane environment can impact the structure and function of integral membrane proteins. As such, elucidation of the thermodynamic driving forces governing protein partitioning between membrane domains of varying lipid composition is a fundamental topic in membrane biophysics. Molecular dynamics simulations provide valuable tools for quantitatively characterizing the free energy landscapes governing protein partitioning at the molecular level. In this study, we propose an efficient simulation methodology for the calculation of free energies for the partitioning of transmembrane proteins between liquid-disorder (Ld) and liquid-ordered (Lo) domains in all-atom (AA) phase-separated lipid bilayers. The computed potential of mean force defining the equilibrium partition coefficients is compared for AA and coarse-grained systems. Energy decomposition is used to identify differences in the underlying thermodynamics. Our findings highlight the importance of employing AA models to accurately estimate relevant free energy changes during protein translation between membrane domains.
众所周知,膜环境会影响整体膜蛋白的结构和功能。因此,阐明支配蛋白质在不同脂质组成的膜域之间分配的热力学驱动力是膜生物物理学的一个基本课题。分子动力学模拟为在分子水平上定量描述支配蛋白质分区的自由能景观提供了宝贵的工具。在本研究中,我们提出了一种高效的模拟方法,用于计算全原子(AA)相分离脂质双层中跨膜蛋白在液态有序(Ld)和液态有序(Lo)结构域之间分区的自由能。比较了 AA 和粗粒度系统中定义平衡分区系数的平均力势的计算结果。能量分解用于确定基本热力学的差异。我们的研究结果强调了采用 AA 模型来准确估计蛋白质在膜域间转化过程中相关自由能变化的重要性。
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
