Federico Droghetti, Florian Lemken, Lubomír Rulíšek, Albert Ruggi, Mirco Natali
{"title":"Selective and Efficient Light-Driven CO2 Reduction to CO with a Heptacoordinated Polypyridine Iron(II) Catalyst","authors":"Federico Droghetti, Florian Lemken, Lubomír Rulíšek, Albert Ruggi, Mirco Natali","doi":"10.1021/acscatal.4c04290","DOIUrl":null,"url":null,"abstract":"The selective generation of carbon-based products in the presence of proton donors currently represents one of the major goals in the catalysis of the CO<sub>2</sub> reduction reaction (CO<sub>2</sub>RR). Within this framework, the iron complex of the 1-([2,2′-bipyridin]-6-yl)-<i>N</i>-([2,2′-bipyridin]-6-ylmethyl)-<i>N</i>-(pyridin-2-ylmethyl) methanamine ligand (DBPy-PyA) turns out to be a selective and efficient catalyst to promote the conversion of CO<sub>2</sub> into CO. In the present work, we report a detailed experimental and computational investigation of the CO<sub>2</sub>RR by this metal complex. Efficient formation of CO (selectivity >90%) was attained under electrochemical conditions (applied potential of −2.0 V vs Fc<sup>+</sup>/Fc) using trifluoroethanol as the proton donor, which provides the best balance, among those tested, in terms of Lewis and Brønsted acidity. This is indeed instrumental in accelerating CO<sub>2</sub> activation while minimizing the parallel generation of hydrogen byproduct. The high activity and selectivity toward CO formation were shown to arise from (i) the ability of the ligand to assist via intramolecular routes the formation of the metallacarboxylic acid catalytic intermediate, (ii) the favorable and almost barrierless detachment of the CO product from the putative iron(II) carbonyl intermediate, and (iii) the weak tendency of the two-electron-reduced complex to form the metal-hydride species. The CO<sub>2</sub>RR by the titled complex was further investigated under light-driven catalytic conditions with [Ru(bpy)<sub>3</sub>]<sup>2+</sup> (bpy = 2,2′-bipyridine) as the sensitizer and <i>N</i>,<i>N</i>-diisopropylethylamine (DIPEA) as the electron donor, leading to unprecedented performances under 1 sun irradiation (0.85 mL CO per mL of solution, quantum yield of 9.4%, selectivity >97%, solely limited by degradation of the sensitizer). Transient absorption spectroscopy suggested that, for the three-component photochemical system examined, catalyst activation by the photogenerated reductant represents the rate-determining step of the photosynthetic process. With this information in hand, by carefully modulating the photon flux, we succeeded in achieving a more than 3-fold enhancement in the quantum yield of CO formation (up to 28%). All in all, our study showcases the great, but often underestimated, potential of molecular catalysis to target efficient and selective transformations.","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"126 1","pages":""},"PeriodicalIF":13.1000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acscatal.4c04290","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
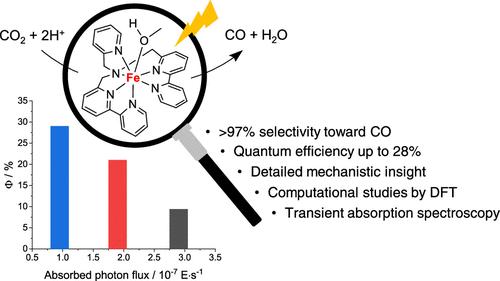
The selective generation of carbon-based products in the presence of proton donors currently represents one of the major goals in the catalysis of the CO2 reduction reaction (CO2RR). Within this framework, the iron complex of the 1-([2,2′-bipyridin]-6-yl)-N-([2,2′-bipyridin]-6-ylmethyl)-N-(pyridin-2-ylmethyl) methanamine ligand (DBPy-PyA) turns out to be a selective and efficient catalyst to promote the conversion of CO2 into CO. In the present work, we report a detailed experimental and computational investigation of the CO2RR by this metal complex. Efficient formation of CO (selectivity >90%) was attained under electrochemical conditions (applied potential of −2.0 V vs Fc+/Fc) using trifluoroethanol as the proton donor, which provides the best balance, among those tested, in terms of Lewis and Brønsted acidity. This is indeed instrumental in accelerating CO2 activation while minimizing the parallel generation of hydrogen byproduct. The high activity and selectivity toward CO formation were shown to arise from (i) the ability of the ligand to assist via intramolecular routes the formation of the metallacarboxylic acid catalytic intermediate, (ii) the favorable and almost barrierless detachment of the CO product from the putative iron(II) carbonyl intermediate, and (iii) the weak tendency of the two-electron-reduced complex to form the metal-hydride species. The CO2RR by the titled complex was further investigated under light-driven catalytic conditions with [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) as the sensitizer and N,N-diisopropylethylamine (DIPEA) as the electron donor, leading to unprecedented performances under 1 sun irradiation (0.85 mL CO per mL of solution, quantum yield of 9.4%, selectivity >97%, solely limited by degradation of the sensitizer). Transient absorption spectroscopy suggested that, for the three-component photochemical system examined, catalyst activation by the photogenerated reductant represents the rate-determining step of the photosynthetic process. With this information in hand, by carefully modulating the photon flux, we succeeded in achieving a more than 3-fold enhancement in the quantum yield of CO formation (up to 28%). All in all, our study showcases the great, but often underestimated, potential of molecular catalysis to target efficient and selective transformations.
目前,在质子供体存在的情况下选择性生成碳基产物是二氧化碳还原反应(CO2RR)催化的主要目标之一。在此框架内,1-([2,2′-联吡啶]-6-基)-N-([2,2′-联吡啶]-6-基甲基)-N-(吡啶-2-基甲基)甲胺配体(DBPy-PyA)的铁配合物被证明是促进 CO2 转化为 CO 的选择性高效催化剂。在本研究中,我们对该金属复合物的 CO2RR 进行了详细的实验和计算研究。在电化学条件下(应用电位为 -2.0 V vs Fc+/Fc),使用三氟乙醇作为质子供体,实现了 CO 的高效生成(选择性达 90%)。这确实有助于加速二氧化碳的活化,同时最大限度地减少氢气副产物的平行生成。研究表明,CO 生成的高活性和高选择性源于:(i) 配体通过分子内途径帮助金属羧酸催化中间体形成的能力;(ii) CO 产物与推定的铁(II)羰基中间体的有利且几乎无障碍的分离;(iii) 双电子还原复合物形成金属酸酐物种的微弱趋势。在光照驱动催化条件下,以[Ru(bpy)3]2+(bpy = 2,2′-联吡啶)为敏化剂,N,N-二异丙基乙胺(DIPEA)为电子供体,进一步研究了标题配合物的 CO2RR。85 mL CO/mL溶液,量子产率为 9.4%,选择性为 97%,仅受敏化剂降解的限制)。瞬态吸收光谱表明,在所研究的三组分光化学系统中,光生还原剂对催化剂的激活是光合作用过程中决定速率的一步。有了这些信息,通过仔细调节光子通量,我们成功地将 CO 形成的量子产率提高了 3 倍多(高达 28%)。总之,我们的研究展示了分子催化在高效和选择性转化方面的巨大潜力,但这一潜力往往被低估了。
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
