{"title":"pH-Mediated Solution-Phase Proton Transfer Drives Enhanced Electrochemical Hydrogenation of Phenol in Alkaline Electrolyte","authors":"Brianna Markunas, Taber Yim, Joshua Snyder","doi":"10.1021/acscatal.4c04874","DOIUrl":null,"url":null,"abstract":"The faradaic efficiency (FE) of the electrochemical hydrogenation (ECH) of phenol and other biomass-derived model compounds could potentially be improved by operating in alkaline electrolytes, where the parasitic hydrogen evolution reaction rate is significantly slower due to a higher Volmer step barrier. However, this approach is potentially limited by the impact of the higher barrier for adsorbed hydrogen (H<sub>ad</sub>) formation, as hydrocarbon ECH is expected to be limited by a hydrogen atom transfer, progressing through a Langmuir–Hinshelwood-type (LH) mechanism. In this work, we show that there are contrasting pH trends for phenol ECH between Pt and Rh, two common catalysts for ECH reactions. Phenol ECH FE and rate on Pt is highest in acidic electrolytes of pH ≤ 5, while activity on Rh is highest near pH 9–10. While our kinetic analysis supports a LH mechanism for Pt at all pH, phenol ECH on Rh shifts from a LH mechanism at low pH to being limited by a direct proton-coupled electron transfer (Eley–Rideal-type mechanism) in which surface adsorbed phenol is hydrogenated by solution-phase H-transfer. We show that the peak activity on Rh at pH 9–10 is due to the proximity of the pH to the p<i>K</i><sub>a</sub> of phenol (p<i>K</i><sub>a</sub> = 10.0). The reversibility of protonation/deprotonation of phenol when electrolyte pH matches its p<i>K</i><sub>a</sub> helps to mediate H-transfer from solution to adsorbed phenol. We also discuss the role of buffer species in mitigating the local pH change and as a H-donor in phenol ECH on Rh at alkaline pH.","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"6 1","pages":""},"PeriodicalIF":13.1000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acscatal.4c04874","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
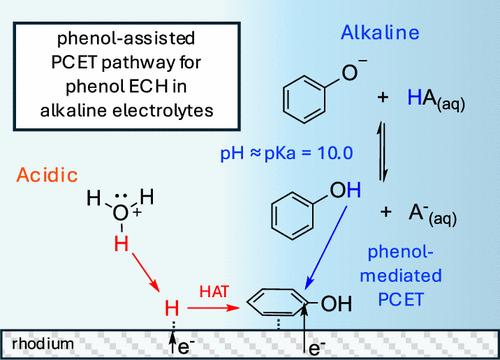
Abstract
The faradaic efficiency (FE) of the electrochemical hydrogenation (ECH) of phenol and other biomass-derived model compounds could potentially be improved by operating in alkaline electrolytes, where the parasitic hydrogen evolution reaction rate is significantly slower due to a higher Volmer step barrier. However, this approach is potentially limited by the impact of the higher barrier for adsorbed hydrogen (Had) formation, as hydrocarbon ECH is expected to be limited by a hydrogen atom transfer, progressing through a Langmuir–Hinshelwood-type (LH) mechanism. In this work, we show that there are contrasting pH trends for phenol ECH between Pt and Rh, two common catalysts for ECH reactions. Phenol ECH FE and rate on Pt is highest in acidic electrolytes of pH ≤ 5, while activity on Rh is highest near pH 9–10. While our kinetic analysis supports a LH mechanism for Pt at all pH, phenol ECH on Rh shifts from a LH mechanism at low pH to being limited by a direct proton-coupled electron transfer (Eley–Rideal-type mechanism) in which surface adsorbed phenol is hydrogenated by solution-phase H-transfer. We show that the peak activity on Rh at pH 9–10 is due to the proximity of the pH to the pKa of phenol (pKa = 10.0). The reversibility of protonation/deprotonation of phenol when electrolyte pH matches its pKa helps to mediate H-transfer from solution to adsorbed phenol. We also discuss the role of buffer species in mitigating the local pH change and as a H-donor in phenol ECH on Rh at alkaline pH.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
