{"title":"First-Principles Study of Structural, Electronic, Magnetic, and Thermodynamic Properties of Tetrataenite L10-FeNi Alloy","authors":"Z. Zine, N. Meftah","doi":"10.1134/S1063783424601115","DOIUrl":null,"url":null,"abstract":"<p>Iron–nickel alloys have received substantial interest because of their exceptional properties and diverse applications in technology and industry. In order to investigate their possible applications, the current research explored the structural, electronic, magnetic, and thermodynamic characteristics of tetrataenite L<span>\\({{1}_{0}}\\)</span>-FeNi alloy through a first-principles approach. The computations were carried out utilizing the density functional theory’s full-potential linearized augmented plane wave. For the electronic exchange-correlation function, we employed the generalized gradient approximation (GGA) and GGA+U (Hubbard potential). The computed lattice parameter and bulk moduli for tetrataenite L<span>\\({{1}_{0}}\\)</span>-FeNi exhibit excellent accord with previously reported data. The formation energy was calculated to be –0.18 eV/f.u. which confirming the structural stability of tetrataenite. The electronic structure revealed that the 3<i>d</i> orbitals of Ni and Fe are major elemental states that contribute to the metallic characteristics of the body-centered tetragonal (bct) L<span>\\({{1}_{0}}\\)</span>-FeNi. Meanwhile, the thermodynamic characters are investigated using the quasi-harmonic Debye mode. The thermal expansion coefficient and the heat capacities are affected simultaneously by the pressure and temperature.</p>","PeriodicalId":731,"journal":{"name":"Physics of the Solid State","volume":"66 10","pages":"416 - 423"},"PeriodicalIF":1.8000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physics of the Solid State","FirstCategoryId":"101","ListUrlMain":"https://link.springer.com/article/10.1134/S1063783424601115","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"PHYSICS, CONDENSED MATTER","Score":null,"Total":0}
引用次数: 0
Abstract
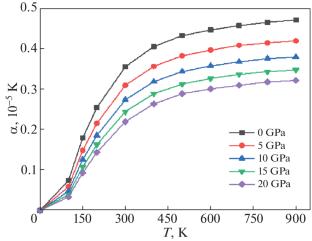
Iron–nickel alloys have received substantial interest because of their exceptional properties and diverse applications in technology and industry. In order to investigate their possible applications, the current research explored the structural, electronic, magnetic, and thermodynamic characteristics of tetrataenite L\({{1}_{0}}\)-FeNi alloy through a first-principles approach. The computations were carried out utilizing the density functional theory’s full-potential linearized augmented plane wave. For the electronic exchange-correlation function, we employed the generalized gradient approximation (GGA) and GGA+U (Hubbard potential). The computed lattice parameter and bulk moduli for tetrataenite L\({{1}_{0}}\)-FeNi exhibit excellent accord with previously reported data. The formation energy was calculated to be –0.18 eV/f.u. which confirming the structural stability of tetrataenite. The electronic structure revealed that the 3d orbitals of Ni and Fe are major elemental states that contribute to the metallic characteristics of the body-centered tetragonal (bct) L\({{1}_{0}}\)-FeNi. Meanwhile, the thermodynamic characters are investigated using the quasi-harmonic Debye mode. The thermal expansion coefficient and the heat capacities are affected simultaneously by the pressure and temperature.
期刊介绍:
Presents the latest results from Russia’s leading researchers in condensed matter physics at the Russian Academy of Sciences and other prestigious institutions. Covers all areas of solid state physics including solid state optics, solid state acoustics, electronic and vibrational spectra, phase transitions, ferroelectricity, magnetism, and superconductivity. Also presents review papers on the most important problems in solid state physics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
