Chunxiao Han, Changshui Chen, Yuxin Zhang, Haibo Li
{"title":"Analysis of clinical phenotypes and genetic variations in two pedigrees affected with Weiss-Kruszka syndrome.","authors":"Chunxiao Han, Changshui Chen, Yuxin Zhang, Haibo Li","doi":"10.1186/s12920-024-02035-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Weiss-Kruszka syndrome (WSKA) is a rare autosomal dominant syndrome characterized by multiple congenital anomalies caused by variants in the zinc finger protein 462 gene (ZNF462). About 40 cases of Weiss-Kruszka syndrome have been reported worldwide, and the aim of this study was to investigate the genetic causes of three patients from two Weiss-Kruszka syndrome family pedigrees with the aim of accumulating more data on the disease.</p><p><strong>Objective: </strong>To explore the clinical and genetic characteristics of two pedigrees with Weiss-Kruszka syndrome.</p><p><strong>Methods: </strong>The clinical data and family history of patients and family members of two pedigrees with Weiss-Kruszka syndrome were collected, and the pathogenic genes of the patients were analysed by whole-exon sequencing. Suspicious variants were verified by Sanger sequencing verification and bioinformatics prediction.</p><p><strong>Results: </strong>Proband 1 has developmental delay, autistic behaviour, and abnormal electroencephalogram results. WES revealed a classical heterozygous c.6696-2 A > C splice variant of the ZNF462 gene, which was detected in neither parent. This position was conserved, and the variant was predicted to be deleterious. Minigene assays revealed that three types of aberrantly spliced mRNAs were produced. MRI of proband 2 suggested dysplasia of the corpus callosum with the formation of hemispheric cleft cysts, with a teardrop-like appearance in the lateral ventricle. WES revealed that a heterozygous c.4891 C > T:p. The Glu1631Ter nonsense variant of the ZNF462 gene was inherited from her mother. According to the guidelines of the American Society of Medical Genetics and combined with its clinical manifestations, c.6696-2 A > C and c.4891 C > T:p. Glu1631Ter was determined to be a possible pathogenic variant.</p><p><strong>Conclusion: </strong>The c.6696-2 A>C and c.4891C > T:p.Glu1631Ter of the ZNF462 gene likely underlies Weiss-Kruszka syndrome in children (foetus), which enriches the variant spectrum of Chinese patients with Weiss-Kruszka syndrome and provides a basis for prenatal diagnosis and genetic counselling.</p>","PeriodicalId":8915,"journal":{"name":"BMC Medical Genomics","volume":"17 1","pages":"261"},"PeriodicalIF":2.0000,"publicationDate":"2024-11-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11536911/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12920-024-02035-x","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Weiss-Kruszka syndrome (WSKA) is a rare autosomal dominant syndrome characterized by multiple congenital anomalies caused by variants in the zinc finger protein 462 gene (ZNF462). About 40 cases of Weiss-Kruszka syndrome have been reported worldwide, and the aim of this study was to investigate the genetic causes of three patients from two Weiss-Kruszka syndrome family pedigrees with the aim of accumulating more data on the disease.
Objective: To explore the clinical and genetic characteristics of two pedigrees with Weiss-Kruszka syndrome.
Methods: The clinical data and family history of patients and family members of two pedigrees with Weiss-Kruszka syndrome were collected, and the pathogenic genes of the patients were analysed by whole-exon sequencing. Suspicious variants were verified by Sanger sequencing verification and bioinformatics prediction.
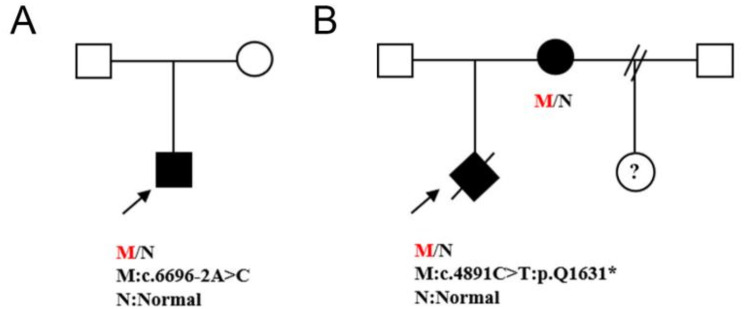
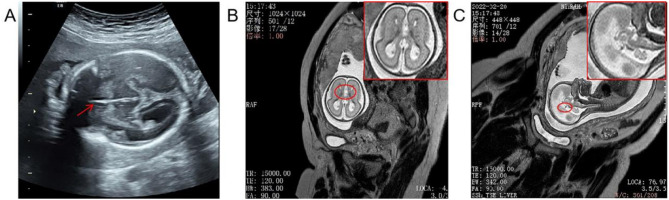
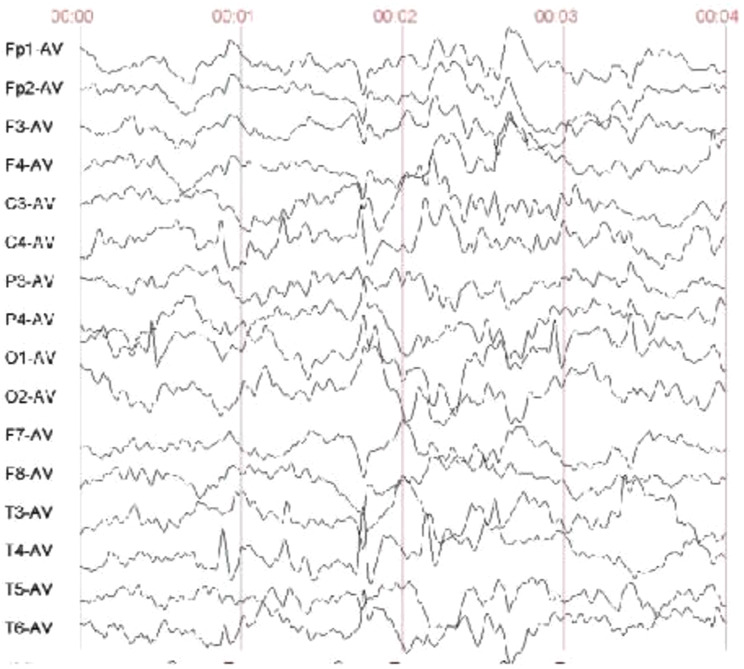
Results: Proband 1 has developmental delay, autistic behaviour, and abnormal electroencephalogram results. WES revealed a classical heterozygous c.6696-2 A > C splice variant of the ZNF462 gene, which was detected in neither parent. This position was conserved, and the variant was predicted to be deleterious. Minigene assays revealed that three types of aberrantly spliced mRNAs were produced. MRI of proband 2 suggested dysplasia of the corpus callosum with the formation of hemispheric cleft cysts, with a teardrop-like appearance in the lateral ventricle. WES revealed that a heterozygous c.4891 C > T:p. The Glu1631Ter nonsense variant of the ZNF462 gene was inherited from her mother. According to the guidelines of the American Society of Medical Genetics and combined with its clinical manifestations, c.6696-2 A > C and c.4891 C > T:p. Glu1631Ter was determined to be a possible pathogenic variant.
Conclusion: The c.6696-2 A>C and c.4891C > T:p.Glu1631Ter of the ZNF462 gene likely underlies Weiss-Kruszka syndrome in children (foetus), which enriches the variant spectrum of Chinese patients with Weiss-Kruszka syndrome and provides a basis for prenatal diagnosis and genetic counselling.
背景:Weiss-Kruszka综合征(WSKA)是一种罕见的常染色体显性遗传综合征,其特征是由锌指蛋白462基因(ZNF462)变异引起的多种先天性畸形。全世界已报道约 40 例 Weiss-Kruszka 综合征病例,本研究旨在调查两个 Weiss-Kruszka 综合征家族谱系中三名患者的遗传原因,以期积累更多有关该病的数据:探讨两个魏氏-克鲁斯卡综合征家系的临床和遗传特征:方法:收集两个魏氏-克鲁斯卡综合征家系中患者及其家庭成员的临床资料和家族史,并通过全外显子测序分析患者的致病基因。通过桑格测序验证和生物信息学预测,对可疑变异进行了验证:结果:前带 1 患有发育迟缓、自闭行为和异常脑电图结果。WES 发现了 ZNF462 基因的经典杂合 c.6696-2 A > C 剪接变异,父母均未检测到该变异。这个位置是保守的,因此该变异被认为是有害的。微型基因检测显示,产生了三种异常剪接的 mRNA。概率 2 的核磁共振成像显示,胼胝体发育不良,形成半球裂囊,侧脑室呈水滴样外观。WES 发现一个杂合子 c.4891 C > T:p。ZNF462基因的Glu1631Ter无义变异遗传自她的母亲。根据美国医学遗传学会(American Society of Medical Genetics)的指南,并结合其临床表现,确定了 c.6696-2 A > C 和 c.4891 C > T:p. Glu1631Ter 无义变体。Glu1631Ter 被确定为可能的致病变异:结论:ZNF462基因的c.6696-2 A>C和c.4891C>T:p.Glu1631Ter可能是儿童(胎儿)魏氏-克鲁兹卡综合征的致病变异,这丰富了中国魏氏-克鲁兹卡综合征患者的变异谱,为产前诊断和遗传咨询提供了依据。
期刊介绍:
BMC Medical Genomics is an open access journal publishing original peer-reviewed research articles in all aspects of functional genomics, genome structure, genome-scale population genetics, epigenomics, proteomics, systems analysis, and pharmacogenomics in relation to human health and disease.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
