Gregory B. Craven, Hang Chu, Jessica D. Sun, Jordan D. Carelli, Brittany Coyne, Hao Chen, Ying Chen, Xiaolei Ma, Subhamoy Das, Wayne Kong, Adam D. Zajdlik, Kin S. Yang, Solomon H. Reisberg, Peter A. Thompson, J. Russell Lipford, Jack Taunton
{"title":"Mutant-selective AKT inhibition through lysine targeting and neo-zinc chelation","authors":"Gregory B. Craven, Hang Chu, Jessica D. Sun, Jordan D. Carelli, Brittany Coyne, Hao Chen, Ying Chen, Xiaolei Ma, Subhamoy Das, Wayne Kong, Adam D. Zajdlik, Kin S. Yang, Solomon H. Reisberg, Peter A. Thompson, J. Russell Lipford, Jack Taunton","doi":"10.1038/s41586-024-08176-4","DOIUrl":null,"url":null,"abstract":"Somatic alterations in the oncogenic kinase AKT1 have been identified in a broad spectrum of solid tumours. The most common AKT1 alteration replaces Glu17 with Lys (E17K) in the regulatory pleckstrin homology domain1, resulting in constitutive membrane localization and activation of oncogenic signalling. In clinical studies, pan-AKT inhibitors have been found to cause dose-limiting hyperglycaemia2–6, which has motivated the search for mutant-selective inhibitors. We exploited the E17K mutation to design allosteric, lysine-targeted salicylaldehyde inhibitors with selectivity for AKT1 (E17K) over wild-type AKT paralogues, a major challenge given the presence of three conserved lysines near the allosteric site. Crystallographic analysis of the covalent inhibitor complex unexpectedly revealed an adventitious tetrahedral zinc ion that coordinates two proximal cysteines in the kinase activation loop while simultaneously engaging the E17K–imine conjugate. The salicylaldimine complex with AKT1 (E17K), but not that with wild-type AKT1, recruits endogenous Zn2+ in cells, resulting in sustained inhibition. A salicylaldehyde-based inhibitor was efficacious in AKT1 (E17K) tumour xenograft models at doses that did not induce hyperglycaemia. Our study demonstrates the potential to achieve exquisite residence-time-based selectivity for AKT1 (E17K) by targeting the mutant lysine together with Zn2+ chelation by the resulting salicylaldimine adduct. A mutant-selective AKT inhibitor shows potential as a targeted therapy for breast cancer, enabling enhanced target engagement and avoiding the dose-limiting toxicity associated with pan-AKT inhibitors.","PeriodicalId":18787,"journal":{"name":"Nature","volume":"637 8044","pages":"205-214"},"PeriodicalIF":48.5000,"publicationDate":"2024-11-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature","FirstCategoryId":"103","ListUrlMain":"https://www.nature.com/articles/s41586-024-08176-4","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
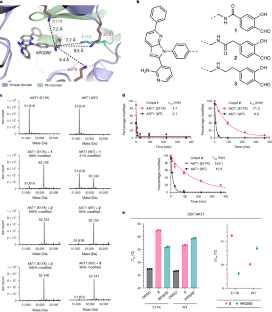
Somatic alterations in the oncogenic kinase AKT1 have been identified in a broad spectrum of solid tumours. The most common AKT1 alteration replaces Glu17 with Lys (E17K) in the regulatory pleckstrin homology domain1, resulting in constitutive membrane localization and activation of oncogenic signalling. In clinical studies, pan-AKT inhibitors have been found to cause dose-limiting hyperglycaemia2–6, which has motivated the search for mutant-selective inhibitors. We exploited the E17K mutation to design allosteric, lysine-targeted salicylaldehyde inhibitors with selectivity for AKT1 (E17K) over wild-type AKT paralogues, a major challenge given the presence of three conserved lysines near the allosteric site. Crystallographic analysis of the covalent inhibitor complex unexpectedly revealed an adventitious tetrahedral zinc ion that coordinates two proximal cysteines in the kinase activation loop while simultaneously engaging the E17K–imine conjugate. The salicylaldimine complex with AKT1 (E17K), but not that with wild-type AKT1, recruits endogenous Zn2+ in cells, resulting in sustained inhibition. A salicylaldehyde-based inhibitor was efficacious in AKT1 (E17K) tumour xenograft models at doses that did not induce hyperglycaemia. Our study demonstrates the potential to achieve exquisite residence-time-based selectivity for AKT1 (E17K) by targeting the mutant lysine together with Zn2+ chelation by the resulting salicylaldimine adduct. A mutant-selective AKT inhibitor shows potential as a targeted therapy for breast cancer, enabling enhanced target engagement and avoiding the dose-limiting toxicity associated with pan-AKT inhibitors.
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
