{"title":"The Bayesian Phylogenetic Bootstrap and its Application to Short Trees and Branches.","authors":"Frédéric Lemoine, Olivier Gascuel","doi":"10.1093/molbev/msae238","DOIUrl":null,"url":null,"abstract":"<p><p>Felsenstein's bootstrap is the most commonly used method to measure branch support in phylogenetics. Current sequencing technologies can result in massive sampling of taxa (e.g. SARS-CoV-2). In this case, the sequences are very similar, the trees are short, and the branches correspond to a small number of mutations (possibly 0). Nevertheless, these trees contain a strong signal, with unresolved parts but a low rate of false branches. With such data, Felsenstein's bootstrap is not satisfactory. Due to the frequentist nature of bootstrap sampling, the expected support of a branch corresponding to a single mutation is ∼63%, even though it is highly likely to be correct. Here, we propose a Bayesian version of the phylogenetic bootstrap in which sites are assigned uninformative prior probabilities. The branch support can then be interpreted as a posterior probability. We do not view the alignment as a small subsample of a large sample of sites, but rather as containing all available information (e.g. as with complete viral genomes, which are becoming routine). We give formulas for expected supports under the assumption of perfect phylogeny, in both the frequentist and Bayesian frameworks, where a branch corresponding to a single mutation now has an expected support of ∼90%. Simulations show that these theoretical results are robust to realistic data. Analyses on low-homoplasy viral and nonviral datasets show that Bayesian bootstrap support is easier to interpret, with high supports for branches very likely to be correct. As homoplasy increases, the two supports become closer and strongly correlated.</p>","PeriodicalId":18730,"journal":{"name":"Molecular biology and evolution","volume":" ","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11600590/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular biology and evolution","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/molbev/msae238","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
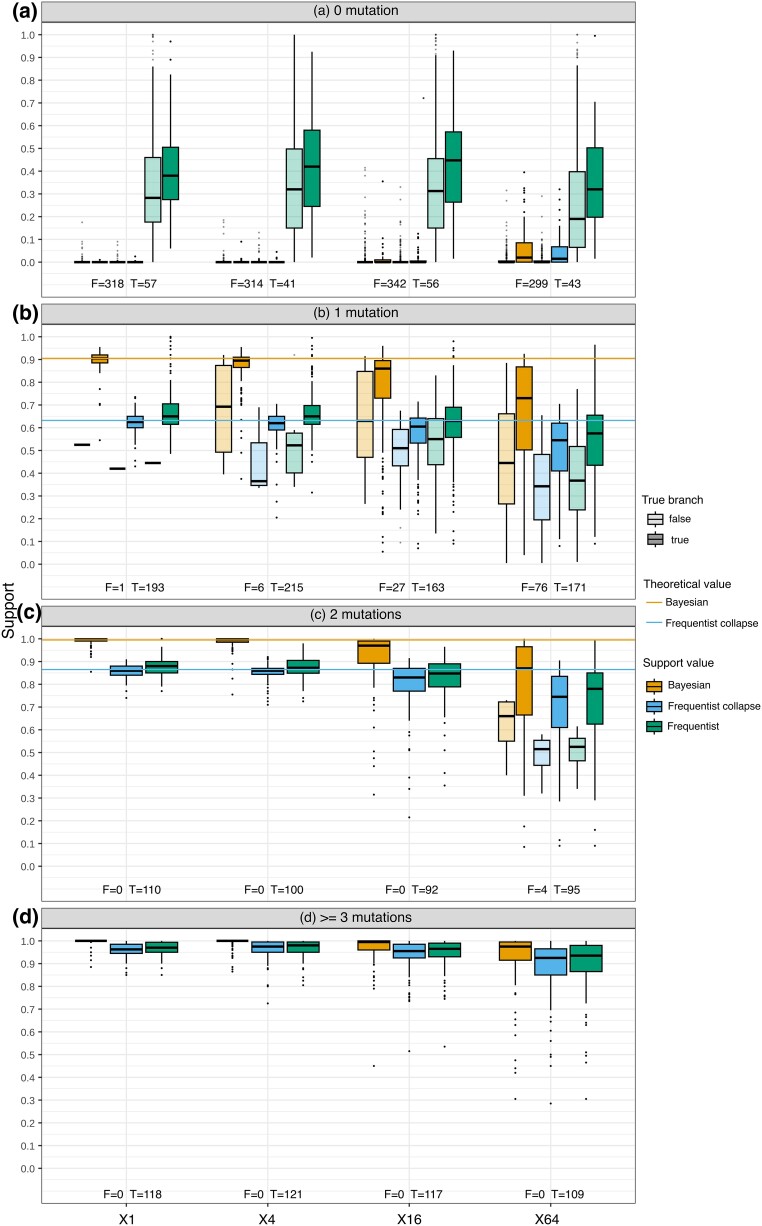
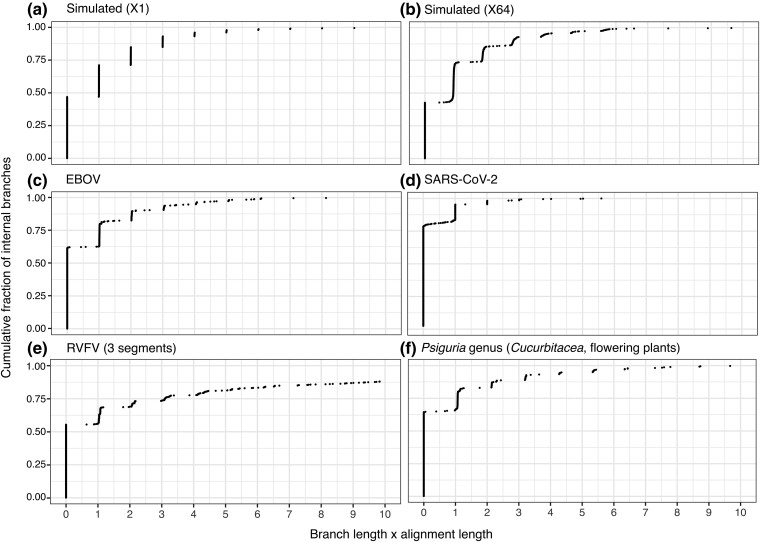
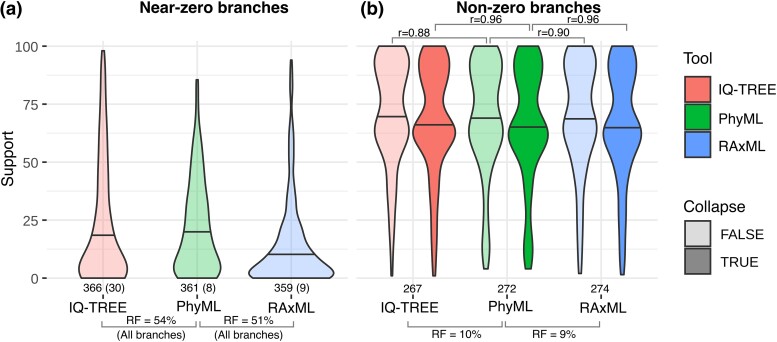
Felsenstein's bootstrap is the most commonly used method to measure branch support in phylogenetics. Current sequencing technologies can result in massive sampling of taxa (e.g. SARS-CoV-2). In this case, the sequences are very similar, the trees are short, and the branches correspond to a small number of mutations (possibly 0). Nevertheless, these trees contain a strong signal, with unresolved parts but a low rate of false branches. With such data, Felsenstein's bootstrap is not satisfactory. Due to the frequentist nature of bootstrap sampling, the expected support of a branch corresponding to a single mutation is ∼63%, even though it is highly likely to be correct. Here, we propose a Bayesian version of the phylogenetic bootstrap in which sites are assigned uninformative prior probabilities. The branch support can then be interpreted as a posterior probability. We do not view the alignment as a small subsample of a large sample of sites, but rather as containing all available information (e.g. as with complete viral genomes, which are becoming routine). We give formulas for expected supports under the assumption of perfect phylogeny, in both the frequentist and Bayesian frameworks, where a branch corresponding to a single mutation now has an expected support of ∼90%. Simulations show that these theoretical results are robust to realistic data. Analyses on low-homoplasy viral and nonviral datasets show that Bayesian bootstrap support is easier to interpret, with high supports for branches very likely to be correct. As homoplasy increases, the two supports become closer and strongly correlated.
期刊介绍:
Molecular Biology and Evolution
Journal Overview:
Publishes research at the interface of molecular (including genomics) and evolutionary biology
Considers manuscripts containing patterns, processes, and predictions at all levels of organization: population, taxonomic, functional, and phenotypic
Interested in fundamental discoveries, new and improved methods, resources, technologies, and theories advancing evolutionary research
Publishes balanced reviews of recent developments in genome evolution and forward-looking perspectives suggesting future directions in molecular evolution applications.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
