Julielson Dos Santos Sousa, Eriosvaldo Florentino Gusmão, Anne Kéllen de Nazaré Dos Reis Dias, Roberto Luiz Andrade Haiduke
{"title":"Relativistic Prolapse-Free Gaussian Basis Sets of Double- and Triple-ζ Quality for <i>s</i>- and <i>p</i>-Block Elements: (aug-)RPF-2Z and (aug-)RPF-3Z.","authors":"Julielson Dos Santos Sousa, Eriosvaldo Florentino Gusmão, Anne Kéllen de Nazaré Dos Reis Dias, Roberto Luiz Andrade Haiduke","doi":"10.1021/acs.jctc.4c01211","DOIUrl":null,"url":null,"abstract":"<p><p>This study presents two new relativistic Gaussian basis sets without variational prolapse of double- and triple-ζ quality, RPF-2Z and RPF-3Z, along with augmented versions including additional diffuse functions, aug-RPF-2Z and aug-RPF-3Z, which are available for all <i>s</i> and <i>p</i> block elements from Hydrogen to Oganesson. The exponents of the Correlation/Polarization (C/P) functions are obtained from a polynomial version of the generator coordinate Dirac-Fock method (p-GCDF). The choice of C/P functions was guided by multireference configuration interaction calculations with single and double excitations (MR-CISD) based on a valence active space. Finally, calculations of fundamental properties done for atomic and molecular systems (bond lengths, vibrational frequencies, dipole moments, and electron affinities) ensure the expected quality of these new basis sets, which may also exhibit some computational efficiency advantages. Additionally, the prolapse-free feature of these sets must provide a reliable description of properties more dependent on core electron distributions, as well.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"9991-9998"},"PeriodicalIF":5.5000,"publicationDate":"2024-11-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11603607/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01211","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/14 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
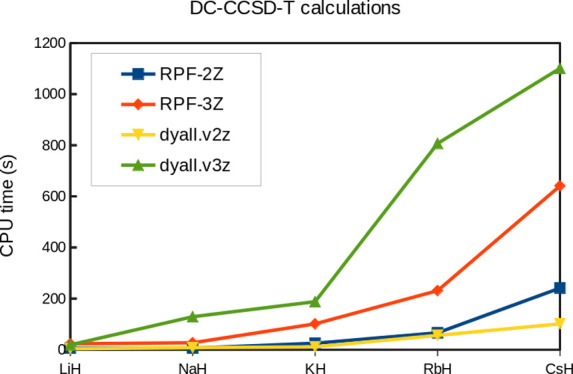
This study presents two new relativistic Gaussian basis sets without variational prolapse of double- and triple-ζ quality, RPF-2Z and RPF-3Z, along with augmented versions including additional diffuse functions, aug-RPF-2Z and aug-RPF-3Z, which are available for all s and p block elements from Hydrogen to Oganesson. The exponents of the Correlation/Polarization (C/P) functions are obtained from a polynomial version of the generator coordinate Dirac-Fock method (p-GCDF). The choice of C/P functions was guided by multireference configuration interaction calculations with single and double excitations (MR-CISD) based on a valence active space. Finally, calculations of fundamental properties done for atomic and molecular systems (bond lengths, vibrational frequencies, dipole moments, and electron affinities) ensure the expected quality of these new basis sets, which may also exhibit some computational efficiency advantages. Additionally, the prolapse-free feature of these sets must provide a reliable description of properties more dependent on core electron distributions, as well.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
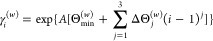
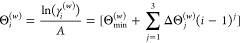



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
