{"title":"DeepMEns: an ensemble model for predicting sgRNA on-target activity based on multiple features.","authors":"Shumei Ding, Jia Zheng, Cangzhi Jia","doi":"10.1093/bfgp/elae043","DOIUrl":null,"url":null,"abstract":"<p><p>The CRISPR/Cas9 system developed from Streptococcus pyogenes (SpCas9) has high potential in gene editing. However, its successful application is hindered by the considerable variability in target efficiencies across different single guide RNAs (sgRNAs). Although several deep learning models have been created to predict sgRNA on-target activity, the intrinsic mechanisms of these models are difficult to explain, and there is still scope for improvement in prediction performance. To overcome these issues, we propose an ensemble interpretable model termed DeepMEns based on deep learning to predict sgRNA on-target activity. By using five different training and validation datasets, we constructed five sub-regressors, each comprising three parts. The first part uses one-hot encoding, wherein 0-1 representation of the secondary structure is used as the input to the convolutional neural network (CNN) with Transformer encoder. The second part uses the DNA shape feature matrix as the input to the CNN with Transformer encoder. The third part uses positional encoding feature matrices as the proposed input into a long short-term memory network with an attention mechanism. These three parts are concatenated through the flattened layer, and the final prediction result is the average of the five sub-regressors. Extensive benchmarking experiments indicated that DeepMEns achieved the highest Spearman correlation coefficient for 6 of 10 independent test datasets as compared to previous predictors, this finding confirmed that DeepMEns can accomplish state-of-the-art performance. Moreover, the ablation analysis also indicated that the ensemble strategy may improve the performance of the prediction model.</p>","PeriodicalId":55323,"journal":{"name":"Briefings in Functional Genomics","volume":" ","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11735754/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in Functional Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bfgp/elae043","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
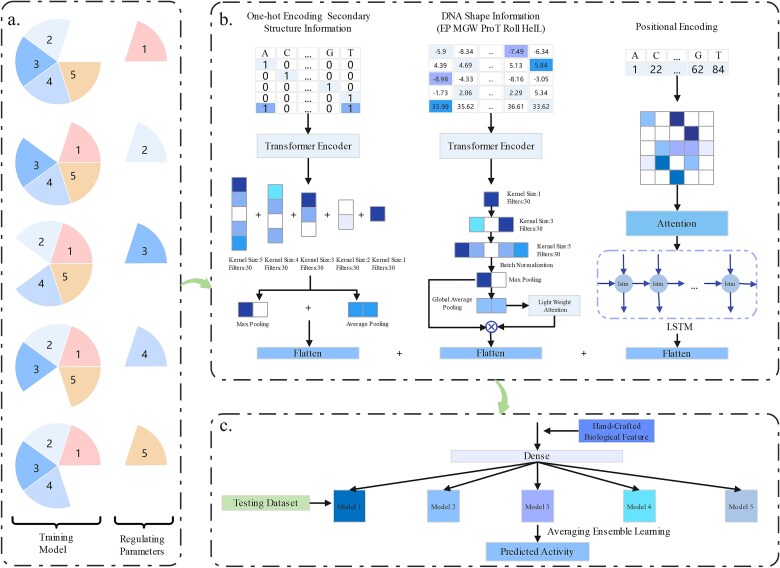
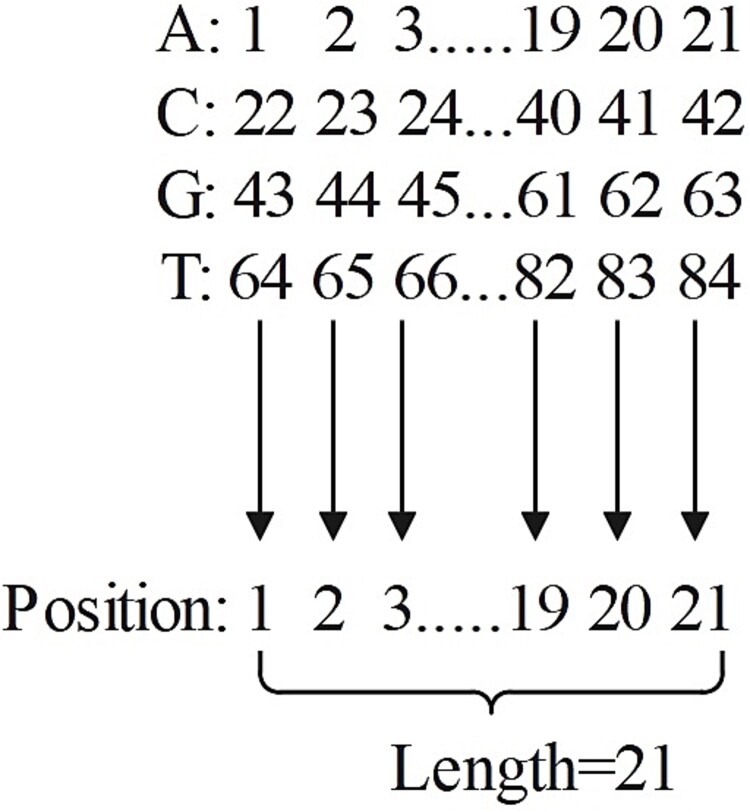
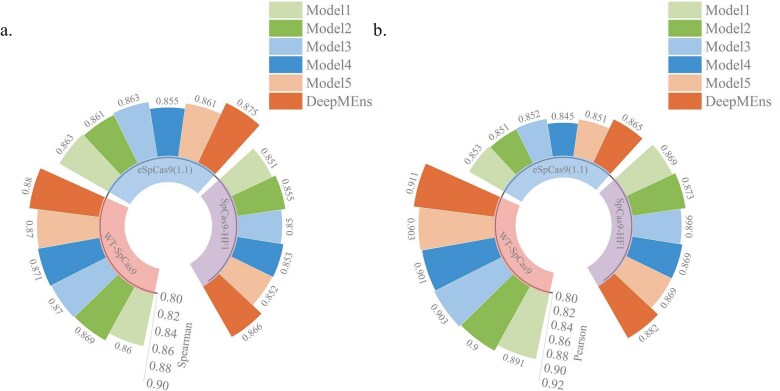
The CRISPR/Cas9 system developed from Streptococcus pyogenes (SpCas9) has high potential in gene editing. However, its successful application is hindered by the considerable variability in target efficiencies across different single guide RNAs (sgRNAs). Although several deep learning models have been created to predict sgRNA on-target activity, the intrinsic mechanisms of these models are difficult to explain, and there is still scope for improvement in prediction performance. To overcome these issues, we propose an ensemble interpretable model termed DeepMEns based on deep learning to predict sgRNA on-target activity. By using five different training and validation datasets, we constructed five sub-regressors, each comprising three parts. The first part uses one-hot encoding, wherein 0-1 representation of the secondary structure is used as the input to the convolutional neural network (CNN) with Transformer encoder. The second part uses the DNA shape feature matrix as the input to the CNN with Transformer encoder. The third part uses positional encoding feature matrices as the proposed input into a long short-term memory network with an attention mechanism. These three parts are concatenated through the flattened layer, and the final prediction result is the average of the five sub-regressors. Extensive benchmarking experiments indicated that DeepMEns achieved the highest Spearman correlation coefficient for 6 of 10 independent test datasets as compared to previous predictors, this finding confirmed that DeepMEns can accomplish state-of-the-art performance. Moreover, the ablation analysis also indicated that the ensemble strategy may improve the performance of the prediction model.
期刊介绍:
Briefings in Functional Genomics publishes high quality peer reviewed articles that focus on the use, development or exploitation of genomic approaches, and their application to all areas of biological research. As well as exploring thematic areas where these techniques and protocols are being used, articles review the impact that these approaches have had, or are likely to have, on their field. Subjects covered by the Journal include but are not restricted to: the identification and functional characterisation of coding and non-coding features in genomes, microarray technologies, gene expression profiling, next generation sequencing, pharmacogenomics, phenomics, SNP technologies, transgenic systems, mutation screens and genotyping. Articles range in scope and depth from the introductory level to specific details of protocols and analyses, encompassing bacterial, fungal, plant, animal and human data.
The editorial board welcome the submission of review articles for publication. Essential criteria for the publication of papers is that they do not contain primary data, and that they are high quality, clearly written review articles which provide a balanced, highly informative and up to date perspective to researchers in the field of functional genomics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
