H. Varela-Rodríguez, A. Guzman-Pando, J. Camarillo-Cisneros
{"title":"Screening and computational characterization of novel antimicrobial cathelicidins from amphibian transcriptomic data","authors":"H. Varela-Rodríguez, A. Guzman-Pando, J. Camarillo-Cisneros","doi":"10.1016/j.compbiolchem.2024.108276","DOIUrl":null,"url":null,"abstract":"<div><div>As cold-blooded organisms living in damp and dark environments, amphibians have evolved robust defense mechanisms to protect themselves from predators and infections. Among the wide repertoire of bioactive compounds they produce are antimicrobial peptides (AMPs), which are required as part of innate immunity. One important class of AMPs is cathelicidins, known for their broad-spectrum activity against pathogens and their immunoregulatory roles. However, despite their promising biomedical potential and the increasing availability of omics data, few cathelicidins have been studied in amphibians, mostly through conventional experimental techniques. Here, we present 210 novel cathelicidin sequences from amphibian transcriptomes, identified through a comprehensive computational pipeline, which employed HMMER and BLAST tools to screen cathelicidin domains. These sequences reveal a typical tripartite domain architecture that was confirmed by SignalP and InterProScan analysis. Phylogenetic inference with IQ-TREE classified the sequences into six categories based on evolutionary relationships. Compared to cathelicidins from other vertebrates, amphibian mature peptides exhibit longer average lengths (around 50 amino acids), fewer aromatic and hydrophobic residues, and reduced thermal stability. Furthermore, these amphibian cathelicidins were characterized for their physicochemical and biological properties, revealing significant antimicrobial potential with lower hemolytic capability, especially in anurans, which suggests a balance between their antimicrobial and hemolytic activities predicted through AMPlify, ampir, AmpGram, and HemoPI. Secondary structure estimations, including three-dimensional modeling using AlphaFold2, indicate that amphibian cathelicidins predominantly feature <span><math><mi>α</mi></math></span>-helices and coils. Some representative models also display a high <span><math><mi>α</mi></math></span>-helix composition with amphipathic topology, facilitating interactions with simulated bacterial membranes as assessed by the PPM approach. Thus, these findings highlight the functional role of cathelicidins in amphibian immunity and their promising biomedical applicability, emphasizing the importance of applying computational methods to expand the scope and reveal the diverse landscape of cathelicidins across vertebrates.</div></div>","PeriodicalId":10616,"journal":{"name":"Computational Biology and Chemistry","volume":"113 ","pages":"Article 108276"},"PeriodicalIF":3.1000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Biology and Chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1476927124002640","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/13 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
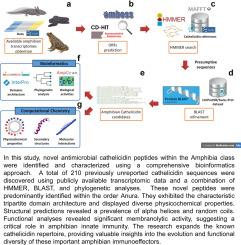
As cold-blooded organisms living in damp and dark environments, amphibians have evolved robust defense mechanisms to protect themselves from predators and infections. Among the wide repertoire of bioactive compounds they produce are antimicrobial peptides (AMPs), which are required as part of innate immunity. One important class of AMPs is cathelicidins, known for their broad-spectrum activity against pathogens and their immunoregulatory roles. However, despite their promising biomedical potential and the increasing availability of omics data, few cathelicidins have been studied in amphibians, mostly through conventional experimental techniques. Here, we present 210 novel cathelicidin sequences from amphibian transcriptomes, identified through a comprehensive computational pipeline, which employed HMMER and BLAST tools to screen cathelicidin domains. These sequences reveal a typical tripartite domain architecture that was confirmed by SignalP and InterProScan analysis. Phylogenetic inference with IQ-TREE classified the sequences into six categories based on evolutionary relationships. Compared to cathelicidins from other vertebrates, amphibian mature peptides exhibit longer average lengths (around 50 amino acids), fewer aromatic and hydrophobic residues, and reduced thermal stability. Furthermore, these amphibian cathelicidins were characterized for their physicochemical and biological properties, revealing significant antimicrobial potential with lower hemolytic capability, especially in anurans, which suggests a balance between their antimicrobial and hemolytic activities predicted through AMPlify, ampir, AmpGram, and HemoPI. Secondary structure estimations, including three-dimensional modeling using AlphaFold2, indicate that amphibian cathelicidins predominantly feature -helices and coils. Some representative models also display a high -helix composition with amphipathic topology, facilitating interactions with simulated bacterial membranes as assessed by the PPM approach. Thus, these findings highlight the functional role of cathelicidins in amphibian immunity and their promising biomedical applicability, emphasizing the importance of applying computational methods to expand the scope and reveal the diverse landscape of cathelicidins across vertebrates.
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
