{"title":"Discovery of a potent anticancer agent against pancreatic ductal adenocarcinoma targeting FAK with DFG-out state and JAK/Aurora kinases","authors":"Rong-Hong Zhang , Ting Chen , Qian-Qian Xiong , Shan Wang , Guo-Qi Chen , Wen-Li Zhang , Hong-Fei Yuan , Yong-Long Zhao , Ting Liu , Yong Huang , Meng Zhou , Cheng-Li Yang , Shang-Gao Liao , Yong-Jun Li","doi":"10.1016/j.ejmech.2024.117059","DOIUrl":null,"url":null,"abstract":"<div><div>Pancreatic ductal adenocarcinoma (PDAC) is a clinically challenging cancer because of the difficulty in diagnosis and its resistance to chemotherapy. Focal adhesion kinase (FAK) is found overexpressed in PDAC, and targeting FAK has been proved to impede the progress of PDAC. However, most of FAK inhibitors were reported to bind with FAK in a DFG-in conformation, leading to a limited anti-tumor effect in clinical studies. Herein, to develop FAK inhibitors targeting the inactive DFG-out conformation, a series of large aromatic rings were selected to improve the interaction with Phe565 of the DFG motif. Compound <strong>26</strong> was designed to effectively inhibit FAK and the proliferation of PANC-1 cells with IC<sub>50</sub> of 50.94 nM and 0.15 μM, respectively. Besides, compound <strong>26</strong> was proved to strongly suppress the proliferation, colony formation, migration, and invasion in FAK-overexpressing PDAC cells. This inhibitor was confirmed to induce the apoptosis and G2/M arrest in PANC-1 cells through the suppression of FAK/PI3K/Akt signal pathway. Meanwhile, compound <strong>26</strong> was found to simultaneously inhibit FAK with DFG-out conformation and JAK3/Aurora B (IC<sub>50</sub> of 9.99 nM and 0.49 nM, respectively). <em>In vivo</em>, compound <strong>26</strong> effectively inhibited the tumorigenesis and metastasis of PDAC with desirable biosafety. Overall, these results suggested that compound <strong>26</strong> was a promising candidate for the treatment of PDAC.</div></div>","PeriodicalId":314,"journal":{"name":"European Journal of Medicinal Chemistry","volume":"282 ","pages":"Article 117059"},"PeriodicalIF":5.9000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0223523424009413","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/15 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
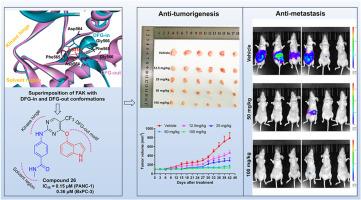
Pancreatic ductal adenocarcinoma (PDAC) is a clinically challenging cancer because of the difficulty in diagnosis and its resistance to chemotherapy. Focal adhesion kinase (FAK) is found overexpressed in PDAC, and targeting FAK has been proved to impede the progress of PDAC. However, most of FAK inhibitors were reported to bind with FAK in a DFG-in conformation, leading to a limited anti-tumor effect in clinical studies. Herein, to develop FAK inhibitors targeting the inactive DFG-out conformation, a series of large aromatic rings were selected to improve the interaction with Phe565 of the DFG motif. Compound 26 was designed to effectively inhibit FAK and the proliferation of PANC-1 cells with IC50 of 50.94 nM and 0.15 μM, respectively. Besides, compound 26 was proved to strongly suppress the proliferation, colony formation, migration, and invasion in FAK-overexpressing PDAC cells. This inhibitor was confirmed to induce the apoptosis and G2/M arrest in PANC-1 cells through the suppression of FAK/PI3K/Akt signal pathway. Meanwhile, compound 26 was found to simultaneously inhibit FAK with DFG-out conformation and JAK3/Aurora B (IC50 of 9.99 nM and 0.49 nM, respectively). In vivo, compound 26 effectively inhibited the tumorigenesis and metastasis of PDAC with desirable biosafety. Overall, these results suggested that compound 26 was a promising candidate for the treatment of PDAC.
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
